
Introduction: Why EMA vs FDA Bioanalytical Method Validation Differences Matter
Understanding EMA vs FDA Bioanalytical Method Validation Differences is extremely important for pharmaceutical companies, CROs, and bioanalytical laboratories that submit studies to both the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Even though both agencies follow similar scientific principles, small regulatory differences can create major issues during submissions. A validation package accepted by one authority may still receive questions, deficiencies, or delays from the other agency if all expectations are not properly addressed.
The main regulatory references are the FDA’s Guidance for Industry: Bioanalytical Method Validation (2018) and the EMA’s Guideline on Bioanalytical Method Validation (2011), along with later EMA Q&A updates. Both guidelines focus on core validation concepts such as accuracy, precision, selectivity, sensitivity, reproducibility, and stability. However, the agencies differ in areas like acceptance criteria, calibration standards, validation design, ISR expectations, and documentation requirements.
This article explains the most important EMA vs FDA Bioanalytical Method Validation Differences in a clear and practical way for bioanalytical scientists, validation specialists, regulatory professionals, and pharmaceutical sponsors involved in global drug development programs.
Explore our specialized services: Bioanalytical Method Development and Validation
Share via:
📋 Article Summary — Key Takeaways
- The FDA Bioanalytical Method Validation Guidance (2018) and the EMA Bioanalytical Method Validation Guideline (2011, including CHMP Q&A updates) have important differences in terminology, validation expectations, acceptance criteria, and required experimental studies.
- EMA treats Incurred Sample Reanalysis (ISR) as a mandatory regulatory activity for pivotal studies, while FDA guidance strongly encourages ISR but allows more scientific flexibility in its implementation.
- EMA applies the term “selectivity” broadly to cover potential matrix-related interferences, whereas the FDA uses both “selectivity” and “specificity” with slightly different practical interpretations during method evaluation.
- Calibration curve acceptance approaches are also different. EMA generally expects at least six of eight non-zero calibration standards to meet acceptance criteria, while the FDA requires a minimum number of acceptable standards without defining the same strict percentage-based rule.
- The agencies also vary in their expectations for QC sample distribution, the number of QC levels included in analytical runs, and the criteria used to determine whether a run passes validation requirements.
- EMA guidance for dried blood spot (DBS) methods and microsampling technologies is currently more detailed and operationally structured, although the FDA has continued to expand its position through draft guidance documents.
- Partial validation and cross-validation requirements are described more specifically by EMA, including clearer guidance on when additional validation studies are necessary after method changes.
- For studies intended for both FDA and EMA submissions, laboratories can develop a single global validation strategy, but the study design should generally follow the more demanding requirement for each validation parameter.
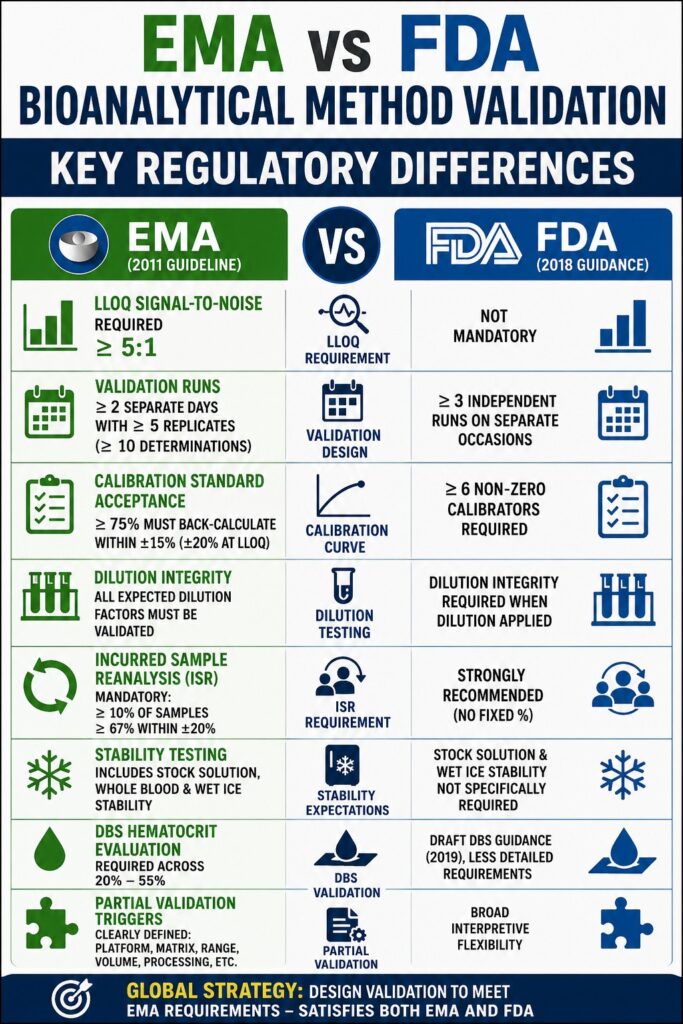
1. Accuracy and Precision Acceptance Criteria: The Main EMA vs FDA Bioanalytical Method Validation Differences
Both the FDA and EMA apply the standard ±15% acceptance criterion for accuracy and precision at most QC levels, with ±20% acceptance at the lower limit of quantification (LLOQ). However, the agencies differ in the required number of validation runs and in how those runs are interpreted during acceptance evaluation.
Number of Validation Runs
FDA (2018)
The FDA recommends at least three independent validation runs performed on separate occasions to demonstrate within-run and between-run accuracy and precision. The guidance expects acceptable performance across runs but provides flexibility in interpretation.
EMA (2011)
The EMA requires validation data generated on at least two separate calendar days with five replicates at each QC concentration level. This results in a minimum of 10 determinations for each QC level. EMA reviewers interpret “separate days” strictly as different calendar days.
LLOQ Acceptance Criteria
Both agencies require:
- Accuracy within ±20%
- Precision not exceeding 20% CV
However, the EMA additionally requires a signal-to-noise ratio of at least 5:1 at the LLOQ. This requirement is not mandatory under FDA guidance.
The FDA focuses more on reproducibility and analytical response, while the EMA requires formal documentation of signal-to-noise performance.
| Parameter | FDA (2018) | EMA (2011) |
|---|---|---|
| Accuracy (non-LLOQ) | ±15% of nominal | ±15% of nominal |
| Accuracy (LLOQ) | ±20% of nominal | ±20% of nominal |
| Precision (non-LLOQ) | ≤15% CV | ≤15% CV |
| Precision (LLOQ) | ≤20% CV | ≤20% CV |
| Minimum validation runs | ≥3 independent runs | ≥2 separate days with ≥5 replicates |
| Minimum determinations | Not specifically defined | ≥10 per QC level |
| LLOQ S/N requirement | Not mandatory | ≥5:1 required |
Need expert support for your next submission? Outsourced Bioanalytical Services
2. Calibration Curve Requirements in EMA vs FDA Bioanalytical Method Validation Differences
The EMA requires that at least 75% of calibration standards successfully back-calculate within ±15% of nominal values, or ±20% at the LLOQ. In practice, this means that a minimum of six out of eight calibration standards must pass.
The FDA requires at least six non-zero calibrators to establish the calibration curve but does not define a fixed minimum percentage of acceptable standards in the same prescriptive manner as the EMA.
Calibration Standard Acceptance
The EMA requires at least 75% of calibration standards to back-calculate within:
- ±15% of nominal values
- ±20% at the LLOQ
This generally means at least six of eight standards must pass.
The FDA requires at least six non-zero calibrators but does not define the same fixed percentage rule as clearly as the EMA.
Anchor Standards
FDA
The FDA allows anchor standards to support calibration curve fitting and modeling. These standards may be used more flexibly.
EMA
The EMA clearly states that anchor standards should not be included in regression calculations and cannot contribute toward calibration acceptance percentages.
This creates an important operational difference. A calibration model acceptable for FDA review may not satisfy EMA expectations if anchor points are used improperly.
Weighting and Statistical Justification
Both agencies require scientific justification for the selected regression model and weighting factor.
However, the EMA generally expects:
- Residual plot analysis
- Statistical comparison between weighting approaches
- Detailed regression evaluation
The FDA also requires justification but allows more flexibility in the level of supporting statistical detail.
Practical Impact
A laboratory using an eight-point LC-MS/MS calibration curve with two anchor standards must ensure that the remaining six calibration standards independently satisfy EMA acceptance requirements. If two of the six fail back-calculation criteria, the EMA would reject the analytical run even if the FDA might still consider the run acceptable depending on the regression approach.
Ensure global compliance for your study: Biomarker Bioanalytical Services for FDA and Health Canada
3. QC Sample Requirements and Analytical Run Acceptance
Both agencies require QC samples at low, medium, and high concentrations in analytical runs. The differences involve QC placement and interpretation of acceptance rules.
| Parameter | FDA (2018) | EMA (2011) |
|---|---|---|
| QC pass threshold | ≥67% within ±15%; ≥50% per level | ≥67% within ±15%; ≥50% per level |
| Minimum QC replicates | Generally 6 across 3 levels | Minimum 4 QCs across ≥3 levels |
| QC placement | Distributed throughout run | Must bracket study samples |
| Dilution QC requirement | Required when dilution applied | Mandatory for all expected dilution factors |
| ULOQ QC requirement | Recommended | Explicitly required |
Dilution Integrity Requirements
EMA
The EMA requires validation of every expected dilution factor separately. For example:
- 1:5 dilution
- 1:10 dilution
Both must be individually validated.
FDA
The FDA supports dilution integrity testing but does not require validation of every dilution factor with the same level of detail.
Because of this, EMA reviewers often examine dilution validation very carefully during inspections and submissions.
Accelerate your early-stage programs: Bioanalytical CRO for Drug Discovery
4. Selectivity and Specificity Requirements
The EMA consistently uses the term “selectivity,” defining it as the method’s ability to differentiate the analyte from endogenous substances, metabolites, and concomitant medications. The FDA uses “selectivity” and “specificity” interchangeably, although the underlying scientific expectation is essentially the same.
Matrix Lot Evaluation
FDA
The FDA recommends testing at least six matrix lots. Hemolyzed and lipemic samples are encouraged when relevant.
EMA
The EMA also requires at least six matrix lots but may additionally expect:
- Hemolyzed matrices
- Lipemic matrices
- Disease-state matrices
This is especially important for studies involving patients with conditions such as renal impairment or hepatic disease.
Concomitant Medication Interference
The EMA explicitly expects interference testing with likely co-administered medications, particularly in combination therapy studies.
The FDA acknowledges the same concern but frames it more as a recommendation than a mandatory validation exercise.
5. Incurred Sample Reanalysis (ISR): One of the Biggest EMA vs FDA Bioanalytical Method Validation Differences
Among all EMA versus FDA bioanalytical method validation differences, ISR requirements often create the greatest operational impact.
EMA ISR Requirements
Under EMA guidance:
- ISR is mandatory for pivotal PK and TK studies
- At least 10% of study samples or a minimum of 10 samples must be reanalyzed
- Samples should include concentrations near Tmax and elimination phase
- At least 67% of ISR results must fall within ±20%
ISR Calculation
∣C1−C2∣(C1+C22)×100\frac{|C1 – C2|}{\left(\frac{C1 + C2}{2}\right)} \times 100(2C1+C2)∣C1−C2∣×100
FDA ISR Requirements
The FDA strongly recommends ISR for pivotal studies but does not officially require a fixed percentage of samples.
The FDA generally allows more scientific flexibility when evaluating ISR failures.
In contrast, EMA reviewers may interpret repeated ISR failures as evidence of poor method reliability.
Recommended Global ISR Strategy
For global studies, laboratories should design ISR programs according to EMA expectations from the beginning. This approach usually satisfies both agencies efficiently.
Support your safety and efficacy trials: Bioanalytical CRO Services for PK and TK
6. Stability Testing Requirements
Both agencies require evaluation of:
- Short-term stability
- Long-term stability
- Freeze-thaw stability
- Processed sample stability
However, the EMA includes more detailed expectations for storage conditions and stock solution stability.
| Stability Type | FDA (2018) | EMA (2011) |
|---|---|---|
| Bench-top stability | Under expected handling conditions | Must exceed maximum handling time |
| Long-term frozen stability | Supports study duration | Must match actual storage conditions |
| Freeze-thaw stability | Minimum 3 cycles | Minimum 3 cycles under study conditions |
| Processed sample stability | Expected autosampler duration | Must exceed typical queue duration |
| Stock solution stability | Recommended | Explicitly required |
| Wet ice stability | Not specifically required | Required if ice storage is used |
| Acceptance criterion | ±15% of fresh QC | ±15% of fresh QC |
Whole Blood Stability
The EMA specifically requires whole blood stability testing whenever blood is collected before plasma separation. Since this is common in clinical PK studies, most laboratories should include whole blood stability in global validation plans.
Maintain the highest quality standards: GLP Bioanalytical Services
7. Microsampling and DBS Validation
The EMA provides more detailed guidance for microsampling and dried blood spot (DBS) validation than the FDA.
EMA DBS Validation Expectations
The EMA requires evaluation of:
- Hematocrit effects
- Volumetric accuracy
- Spot homogeneity
- On-card stability
- DBS-to-plasma correlation
Specific EMA DBS Requirements
Key expectations include:
- Hematocrit testing between 20% and 55%
- Uniform analyte distribution assessment
- Comparative recovery studies
- Stability testing under multiple storage conditions
The FDA published draft DBS guidance in 2019, but EMA expectations remain more detailed and operationally strict.
8. Partial Validation and Cross-Validation Requirements
The EMA provides explicit definitions for scenarios requiring partial validation or cross-validation, whereas the FDA offers broader interpretive flexibility.
EMA Partial Validation Triggers
Partial validation is expected when there are:
- Changes in analytical platform
- Matrix changes
- Anticoagulant changes
- Modified concentration ranges
- Reduced sample volumes
- Significant processing modifications
Cross-Validation Requirements
EMA
The EMA requires cross-validation using at least three QC levels with acceptance based on ±15% mean bias.
FDA
The FDA also requires cross-validation but provides more flexibility regarding QC sample design.
For multi-site studies and global CRO programs, EMA expectations are generally more demanding.
Optimize your development timeline: Bioanalytical Validation Timeline
9. Endogenous Compounds and Ligand Binding Assays
EMA guidance is generally more detailed for endogenous analytes and ligand binding assays, particularly regarding surrogate matrices and parallelism evaluation.
Surrogate Matrix Requirements
Both agencies allow surrogate matrices when endogenous analytes cannot be completely removed from biological samples.
However, the EMA additionally requires:
- Parallelism testing
- Comparison between surrogate matrices and incurred samples
Ligand Binding Assays (LBAs)
The EMA provides detailed expectations for:
- Hook effect assessment
- Minimum dilution requirements
- Parallelism studies
The FDA discusses these topics more generally and allows greater scientific interpretation.
Navigate complex drug modalities with ease: Advanced Bioanalytical Strategies for Complex Drug Modalities
10. Building a Global Validation Strategy for EMA and FDA Submissions
A harmonized validation strategy is achievable when validation protocols are designed according to the stricter requirement for each parameter.
| Parameter | Stricter Standard | Recommended Global Approach |
|---|---|---|
| LLOQ signal-to-noise | EMA | Document ≥5:1 |
| ISR requirements | EMA | Perform mandatory 10% ISR |
| Stock solution stability | EMA | Include in all validations |
| Whole blood stability | EMA | Always evaluate when relevant |
| Calibration anchor handling | EMA | Exclude anchors from regression |
| Hemolyzed/lipemic matrices | EMA | Evaluate target populations |
| Partial validation triggers | EMA | Follow EMA definitions |
| Calibration acceptance | EMA | Apply ≥75% passing rule |
| Dilution integrity | EMA | Validate all dilution factors |
| DBS hematocrit evaluation | EMA | Test across 20–55% hematocrit |
At ResolveMass Laboratories Inc., bioanalytical validation programs are designed using a dual-compliance strategy from the beginning. This helps ensure that studies conducted in Canada can support both FDA and EMA submissions without the need for major re-validation work later. Continuous monitoring of EMA updates, CHMP Q&A revisions, and FDA guidance changes helps maintain globally compliant validation procedures.
Partner with a leader in regulatory science: Bioanalytical CRO Partnership
Conclusion: Why Understanding EMA vs FDA Bioanalytical Method Validation Differences Is Important
The EMA vs FDA Bioanalytical Method Validation Differences discussed in this article are not minor regulatory details. These differences directly affect submission quality, regulatory timelines, study approvals, and overall project success.
From ISR requirements and stock solution stability to calibration curve acceptance and DBS hematocrit assessment, the EMA generally applies a more detailed and prescriptive framework than the FDA.
For pharmaceutical companies, CROs, and bioanalytical laboratories involved in global drug development, the best approach is usually to design studies according to EMA expectations from the beginning. In most cases, this strategy also satisfies FDA requirements and reduces the risk of additional validation work later.
Ready to discuss your bioanalytical strategy? Bioanalytical Strategy for Drug Development
Although ICH M10 harmonization efforts have reduced some historical gaps, several important EMA vs FDA Bioanalytical Method Validation Differences still remain. A strong understanding of these regulatory expectations is essential for organizations working in modern bioanalysis, clinical pharmacology, and regulatory science.
Frequently Asked Questions:
EMA does not automatically accept a method simply because it was validated according to FDA guidance. The validation package must also meet EMA-specific expectations such as ISR compliance, stock solution stability evaluation, LLOQ signal-to-noise ratio requirements, and proper calibration handling. If these details are missing, EMA reviewers may request additional studies or partial re-validation before accepting the data.
EMA considers Incurred Sample Reanalysis (ISR) mandatory for pivotal pharmacokinetic and toxicokinetic studies. The FDA strongly encourages ISR for important clinical studies, especially Phase I and pivotal trials, but uses more flexible wording in its guidance. For global submissions, laboratories usually follow EMA ISR expectations because this approach also satisfies FDA requirements.
According to EMA guidance, at least 67% of ISR samples must show results within ±20% difference between the original value and the repeat analysis. The calculation compares the difference relative to the average of both measurements. If ISR performance fails, EMA may request a detailed root-cause investigation and could question the reliability of the bioanalytical data.
Both agencies use similar QC acceptance rules, including the requirement that at least 67% of QC samples pass within defined limits. However, EMA places stronger emphasis on the placement of QC samples throughout the analytical run. QC samples should bracket study samples across the sequence, while FDA guidance allows laboratories more flexibility in run design.
Both FDA and EMA expect testing of at least six independent matrix lots during selectivity evaluation. However, EMA also expects additional assessments for hemolyzed, lipemic, and disease-state matrices when these conditions may occur in the study population. FDA treats these evaluations more as recommendations instead of strict requirements.
Yes, there is a significant regulatory difference regarding anchor standards. EMA does not allow anchor standards to be included in calibration curve regression calculations. In contrast, FDA guidance is more flexible and may allow their use depending on the method design. For studies intended for both agencies, laboratories generally exclude anchor standards from regression models to maintain EMA compliance.
EMA specifically expects whole blood stability testing to cover the time between blood collection and plasma or serum separation. This includes evaluating storage conditions such as room temperature and ice handling to confirm analyte stability during routine sample processing. FDA also recognizes the importance of whole blood stability, but EMA reviewers usually evaluate this area more strictly during submissions.
EMA provides more detailed DBS validation expectations compared with FDA guidance. EMA requires hematocrit effect studies, spot homogeneity testing, on-card stability assessments, and comparison studies between DBS samples and traditional matrices. Although FDA published draft DBS guidance, EMA requirements remain more comprehensive for international regulatory submissions.
Reference:
- Guideline IH. Bioanalytical method validation M10. European Medicines Agency: Amsterdam, The Netherlands. 2019 Feb.https://www.wrib.org/PDFs/ICH_M10_BMV_Draft_Guideline-190227.pdf
- Gu M, Gehman A, Nifong B, Mayer AP, Li V, Birchler M, Wang K, Tang H. From guidelines to implementation: a case study on applying ICH M10 for bioanalytical assay cross-validation. The AAPS Journal. 2025 Feb 28;27(2):54.https://link.springer.com/article/10.1208/s12248-025-01038-5
- Timmerman P, White S, Adcock N, Arfvidsson C, Barfield M, Cowan K, Ferrari L, Golob M, Goodwin L, Hughes R, Ivanova T. Feedback from a workshop by the European Bioanalysis Forum on assay validation requirements for in vitro assays following the publication of ICH M12 guideline–a plea for context-of-use over ICH M10 standards.https://www.tandfonline.com/doi/abs/10.1080/17576180.2025.2468596

