
Navigating Clinical Translation of CNS Small Molecules Under FDA Fast Track
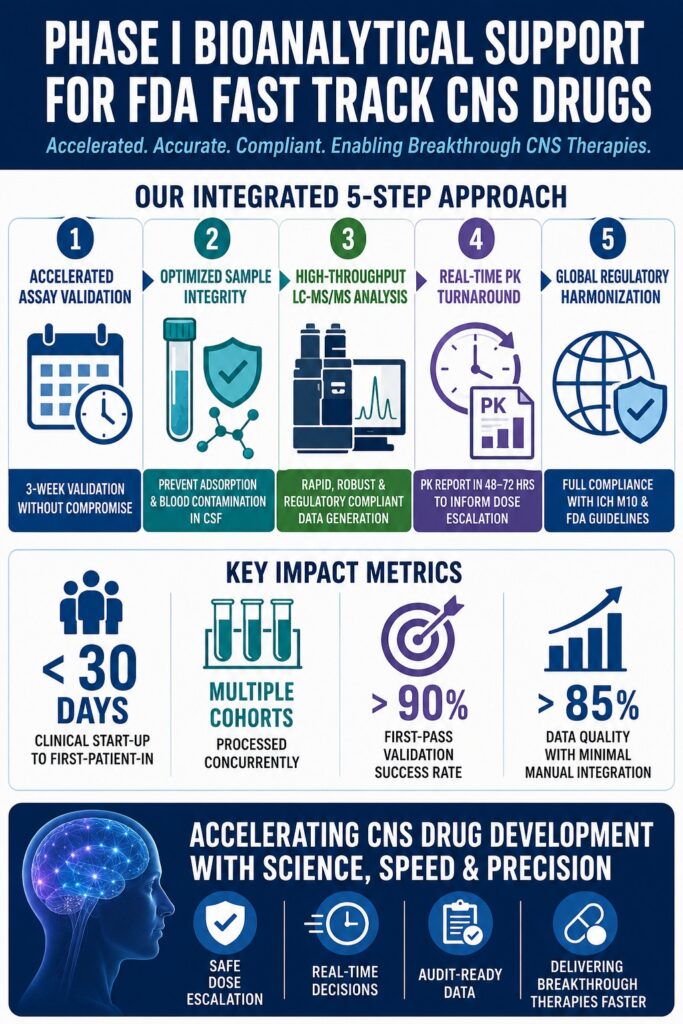
The clinical translation of central nervous system (CNS)-active small molecules under FDA Fast Track pathways requires a significant reduction in conventional clinical start-up and dose-escalation timelines, often compressing schedules to as little as four weeks to satisfy the demands of accelerated drug development. This regulatory acceleration is supported through frequent interactions with the FDA and rolling reviews of completed nonclinical and bioanalytical datasets. As a result, early-phase data must be generated, finalized, and audited within exceptionally short timeframes.
During the clinical advancement of CNS-active drug candidates, establishing a highly coordinated pharmacokinetic (PK) and safety assessment framework is essential. Delivering effective Phase I Bioanalytical Support for Fast Track FDA programs requires the rapid implementation of ultrasensitive quantitative assays capable of characterizing complex compartmental drug distribution while maintaining compliance with condensed clinical timelines. This case study outlines the clinical and technical coordination required to support an investigational CNS small molecule progressing through early-stage human clinical trials under expedited regulatory review.
To learn more about navigating expedited regulatory pathways, visit our Biomarker & Bioanalytical Services for FDA and Health Canada Compliance page.
The primary goal of a first-in-human (FIH) clinical program involving a CNS small molecule is the rapid and safe determination of the tolerated dose range while confirming adequate blood-brain barrier (BBB) penetration. Although preclinical animal studies provide initial dose-scaling estimates, real-time human PK data remains the definitive factor guiding dose-escalation decisions. Successfully translating these compounds into clinical development requires specialized Phase I units operating with hybrid intensive care unit (ICU)-level capabilities, where blood and cerebrospinal fluid (CSF) sampling must be performed with minute-level accuracy to capture the rapid absorption, distribution, and elimination kinetics characteristic of small molecule therapeutics.
Maintaining clinical momentum requires seamless collaboration among the clinical trial site, bioanalytical laboratory, and pharmacokinetic modeling team. Since Fast Track clinical protocols are frequently designed as adaptive studies incorporating both Single Ascending Dose (SAD) and Multiple Ascending Dose (MAD) components, bioanalytical assays must be fully validated and operational before administration of the first dose. This level of preparedness is achieved through parallel method development and validation strategies that allow multiple activities to proceed simultaneously.
Explore specialized early-phase support options at our Bioanalytical CRO for Drug Discovery & Early Phase Support section.
Article Summary:
- FDA Fast Track programs for CNS small molecules significantly shorten traditional clinical development timelines, requiring bioanalytical data generation, validation, and review within weeks rather than months.
- Successful Phase I bioanalytical support depends on rapid assay development, accelerated validation strategies, and close coordination between clinical sites, bioanalytical laboratories, and pharmacokinetic (PK) modeling teams.
- Validation timelines can be reduced from several months to approximately three weeks by conducting multiple validation activities in parallel while maintaining compliance with FDA guidance and ICH M10 requirements.
- Low-protein matrices such as cerebrospinal fluid (CSF) present unique analytical challenges, including nonspecific adsorption of lipophilic compounds and blood contamination during lumbar puncture procedures, both of which can compromise data accuracy if not properly controlled.
- Advanced mitigation strategies, including surfactant-treated collection materials, protein supplementation, biocompatible LC hardware, immediate sample processing, and hemolysis-specific validation studies, help ensure reliable quantification of CNS drug concentrations.
- Adaptive Single Ascending Dose (SAD) and Multiple Ascending Dose (MAD) study designs rely on rapid LC-MS/MS analysis and real-time pharmacokinetic modeling to provide dose-escalation decisions within 48–72 hours of sample analysis.
- Adherence to harmonized regulatory standards such as ICH M10, including incurred sample reanalysis, stability assessments, and rigorous quality controls, enables the generation of submission-ready datasets that support accelerated regulatory review and efficient advancement of promising CNS therapies.
Overcoming Validation Bottlenecks in Phase I Bioanalytical Support for Fast Track FDA
Validation bottlenecks within accelerated clinical programs are addressed by conducting critical assay validation activities in parallel while utilizing validated surrogate matrices. This structured strategy safely reduces traditional validation timelines of four to twelve weeks into an accelerated three-week period without compromising regulatory compliance.
Historically, bioanalytical assay validation follows a sequential workflow in which stability assessments, selectivity studies, and calibration reproducibility evaluations are conducted over several weeks. Under a Fast Track development model, however, this timeline must be substantially compressed.
To achieve a three-week validation schedule in accordance with ICH M10 and FDA guidance, a highly coordinated validation plan is implemented. While the regulatory requirement for three independent accuracy and precision runs conducted across at least three separate days remains mandatory, secondary validation parameters can be evaluated concurrently.
| Validation Experiment | Standard Timeline | Accelerated Timeline | Operational Integration Strategy |
|---|---|---|---|
| Method Development & Optimization | 2–4 Weeks | Pre-Project Phase | Finalize extraction chemistries and MS/MS parameters before formal validation begins. |
| Selectivity, Specificity & Matrix Effects | 3 Days | Days 1–2 | Simultaneously evaluate blank human plasma, blank CSF, hemolyzed lots, and hyperlipidemic lots. |
| Calibration & QC Preparation | 3–5 Days | Days 3–4 | Prepare bulk single-use aliquots of calibration standards and Quality Controls to minimize inter-run preparation variability. |
| Accuracy & Precision (A&P) Runs | 1–3 Weeks | Days 5–7 | Perform the three mandatory validation runs on consecutive days using parallel qualified LC-MS/MS systems. |
| Short-Term & Autosampler Stability | 1–2 Weeks | Days 8–12 | Conduct freeze-thaw, bench-top, and processed sample stability studies concurrently with ongoing matrix evaluations. |
| Dilution Integrity & Recovery | 2–3 Days | Days 13–14 | Verify above-range dilution factors and extraction recovery across low, mid, and high QC concentrations. |
| Long-Term Stability Initiation | 2–6 Weeks | Parallel Phase | Store long-term stability samples at -20°C and -80°C for evaluation during ongoing clinical analysis. |
| Data Analysis & Report Writing | 1–2 Weeks | Days 15–21 | Utilize automated report templates and perform concurrent Quality Assurance (QA) review activities. |
The success of this compressed timeline depends heavily on the pre-validation phase. Any modification to the analytical method during active validation can result in immediate validation failure, effectively resetting the regulatory timeline and introducing significant delays. By establishing a fully optimized and robust extraction procedure before validation begins, first-pass validation success rates can exceed 85%, thereby minimizing costly setbacks.
Learn more about rapid regulatory adherence by checking our guide on Bioanalytical Method Development and Validation Services.
Operational Metrics in Phase I Bioanalytical Support for Fast Track FDA
Operational performance metrics in early-phase clinical programs place significant emphasis on clinical start-up speed, concurrent cohort processing capacity, and data quality measurement. Reducing the time between participant dosing, sample extraction, and PK interpretation is essential to prevent interruptions in trial progression. To sustain accelerated development timelines, clinical units and bioanalytical contract research organizations (CROs) are assessed using highly integrated performance indicators.
Start-up speed is measured as the interval between final protocol approval and first-patient-in (FPI). While conventional timelines frequently exceed 60 days, Fast Track programs typically require FPI within fewer than 30 days. Achieving this objective requires bioanalytical laboratories to maintain pre-approved standard operating procedures (SOPs) for rapid procurement of reference standards, synthesis of custom internal standards, and immediate allocation of analytical instrumentation.
Throughput capacity measures the laboratory’s ability to support multiple clinical cohorts simultaneously. In adaptive trial designs, MAD cohorts often begin before final SAD cohorts are completed, resulting in concurrent arrivals of large volumes of plasma and CSF samples. To manage this workload efficiently, laboratories must maintain dedicated instrument schedules and cross-trained analytical personnel capable of handling overlapping extraction and analysis workflows without creating data backlogs.
For specialized pharmacokinetic operations, review our Bioanalytical CRO Services for PK and TK Analysis.
The data quality index tracks protocol deviations, sample collection timing adherence, and the frequency of manual chromatographic integration. Regulatory auditors reviewing Fast Track submissions closely examine manual integrations. Reducing the need for manual intervention requires highly optimized chromatographic separation capable of fully resolving endogenous isobaric matrix components from the target analyte. This ensures that automated peak integration remains accurate, consistent, and reproducible across thousands of clinical samples.
Discover strategies for balancing speed and cost on our Cost-Effective Bioanalytical Services & Outsourcing Strategies resource page.
Resolving Nonspecific Adsorption of Hydrophobic Analytes in Low-Protein Matrices
Nonspecific adsorption in cerebrospinal fluid can be mitigated by pre-treating clinical collection containers and analytical hardware with non-ionic surfactants or protein-based competitors that saturate hydrophobic surfaces. These surface-blocking agents prevent the physical loss of hydrophobic small molecules and help maintain extraction recoveries above 90%.
Hydrophobic small molecules targeting CNS receptors frequently exhibit high lipophilicity, reflected by elevated log D or log P values, along with limited water solubility. When these compounds enter low-protein environments such as CSF, which lacks the substantial albumin and lipid concentrations found in plasma, they tend to partition out of the aqueous phase and adsorb onto hydrophobic surfaces including collection tubes, pipette tips, storage vials, and chromatographic system components.
Nonspecific adsorption (NSA), also referred to as nonspecific binding (NSB), represents a significant but often unnoticed source of analytical error during clinical PK determination. If left unaddressed, NSA can substantially underestimate drug concentrations, particularly near the lower limit of quantitation (LLOQ), where the ratio of container surface area to analyte concentration is greatest. This phenomenon can produce non-linear calibration curves, artificially reduced recovery values, and poor inter-day precision.
To systematically address NSA during clinical sample collection and laboratory processing, several physical and chemical interventions are implemented:
Pre-Analytical Surfactant Addition
Clinical collection tubes may be pre-treated with non-ionic surfactants such as Tween-20 (0.02% to 0.05%) or Triton X-100. These compounds rapidly assemble at the solid-liquid interface, creating a protective layer that prevents lipophilic analytes from interacting with polypropylene surfaces. Since surfactants can significantly alter electrospray ionization (ESI) performance through ion suppression or enhancement, sample preparation methods must incorporate selective solid-phase extraction (SPE) or liquid-liquid extraction (LLE) procedures to remove surfactants before analytical injection.
Protein Competitor Spiking
Immediately supplementing CSF samples with 0.15% Bovine Serum Albumin (BSA) or human serum albumin (HSA) provides another effective strategy. The added protein binds lipophilic analytes, maintaining their solubility and creating a matrix environment more closely resembling plasma.
PEEK and Hybrid Hardware Surface Deployment
Within liquid chromatography systems, analytes may adsorb to stainless steel tubing, column frits, and injection needles. Replacing conventional stainless-steel components with biocompatible polyether ether ketone (PEEK) tubing or advanced hybrid organic/inorganic surface technologies, such as MaxPeak High Performance Surfaces, eliminates metal-analyte interactions. Published studies have demonstrated that replacing standard stainless-steel frits with hybrid surface technologies can improve analyte recovery from 69% to nearly 100%.
To read about advanced platform techniques, visit our LC-MS/MS Bioanalytical Services & High-Throughput Platforms overview.
Addressing Lumbar Puncture Blood Contamination and Hemolysis in Cerebrospinal Fluid
Blood contamination resulting from lumbar puncture procedures is managed through immediate cold-temperature centrifugation at the clinical site to separate intact cells before hemolysis can occur. This pre-analytical control is complemented by bioanalytical validation studies evaluating assay selectivity and matrix effects in CSF samples intentionally spiked with hemolyzed whole blood.
CSF collection is typically performed through lumbar puncture procedures, during which minor vascular injury occurs in up to 20% of cases. This can introduce peripheral red blood cells and plasma proteins into the CSF sample.
Once red blood cells enter the CSF environment, they become highly susceptible to lysis because of osmotic differences between CSF and whole blood. Hemolysis releases intracellular constituents, including hemoglobin and active enzyme systems such as proteases and esterases, directly into the sample matrix. These enzymes can rapidly degrade drug analytes, while the release of heme significantly alters sample composition. During LC-MS/MS analysis, such contamination may cause severe electrospray ion suppression, negatively affecting assay accuracy and selectivity.
To control these variables, bioanalytical laboratories must validate assay performance in the presence of blood contamination and establish rigorous sample-handling SOPs.
Immediate Clinical Site Centrifugation
Clinical personnel are instructed to centrifuge all CSF samples immediately after collection, for example at 3000 × g for 10 minutes at 4°C, to pellet intact cellular material before lysis occurs. The resulting supernatant is promptly separated, aliquoted, and snap-frozen in liquid nitrogen before long-term storage at -80°C.
Selectivity Validation in Hemolyzed CSF
During method validation, selectivity assessments are performed using blank human CSF samples spiked with 1% to 2% hemolyzed whole blood. These samples are analyzed at the LLOQ and compared against non-hemolyzed CSF controls. The method is considered robust when accuracy remains within 85% to 115% of nominal concentrations and precision demonstrates a %CV of ≤15%.
Because authentic clinical CSF is a limited resource, artificial cerebrospinal fluid (aCSF) is commonly used during development to reduce sample consumption.
| Surrogate Matrix Composition | Target Analyte Physicochemical Profile | Advantages in Method Development | Limitations & Disadvantages |
| 0.15% Bovine Serum Albumin (BSA) in Perfusion Fluid | Hydrophobic/Lipophilic Compounds (High log D) | Mimics natural albumin binding and reduces nonspecific adsorption. | High molecular-weight proteins may accumulate on the LC-MS/MS source if precipitation is incomplete. |
| 0.5% to 2.0% Rat Plasma in Perfusion Fluid | Peptides and Complex Small Molecules | Provides a reproducible matrix with endogenous lipids and proteins for recovery assessment. | May introduce species-specific interferences and active enzymatic systems. |
| Phosphate-Buffered Saline (PBS) / Saline Solutions | Polar/Hydrophilic Compounds (Low log D) | Simple, economical, and free of protein-based interferences. | Provides no protection against nonspecific adsorption of lipophilic analytes. |
| Pure Water / Organic Solvents | Highly Polar and Water-Soluble Compounds | Eliminates matrix-related ion suppression. | Lacks buffering capacity and may produce unstable chromatographic retention for ionizable compounds. |
Explore specialized asset risk reduction metrics on our Bioanalytical Stability Testing & Integrity Assays page.
Synchronizing SAD/MAD Dosing Cohorts with Real-Time PK Turnaround
Synchronization of dosing cohorts is achieved through a rolling bioanalytical processing schedule lasting three to four days, followed by an internal non-compartmental pharmacokinetic modeling cycle completed within 48 to 72 hours. This integrated workflow provides the clinical Safety Review Committee with timely exposure data necessary to authorize dose escalation safely.
Modern early-phase drug development frequently combines Single Ascending Dose (SAD) and Multiple Ascending Dose (MAD) cohorts within adaptive protocols, reducing overall clinical development timelines by as much as six months.
In a typical SAD design, a small group of healthy volunteers, usually eight participants randomized in a 3:1 active-to-placebo ratio, receives a single dose of the investigational drug. To maximize participant safety, protocols often implement sentinel dosing strategies in which two individuals, one receiving active treatment and one receiving placebo, are dosed first and monitored for 48 hours. If no significant adverse events occur, the remaining participants are subsequently dosed.
Following completion of dosing, blood and serial CSF samples are collected over a 24- to 72-hour period to capture the complete pharmacokinetic profile. Samples are shipped to the bioanalytical laboratory on a rolling basis. To meet the stringent timelines established by the Safety Review Committee (SRC), extraction and LC-MS/MS analysis must be completed within three to four days.
After analytical completion, quality-control-reviewed concentration data is transferred to the pharmacokinetic modeling group. Using Phoenix WinNonlin, pharmacokinetic scientists perform non-compartmental analysis (NCA) to calculate critical parameters including:
NCA Parameters Calculated: Cmax, Tmax, AUC0-t, t1/2, λz, Vz/F, and CL/F.
This analysis provides answers to four key questions:
Exposure Levels
What maximum concentration (Cmax) and total systemic exposure (AUC) were achieved at the current dose level?
Absorption and Elimination
How rapidly is the drug absorbed and eliminated from systemic circulation and CNS compartments, as reflected by Tmax and t1/2?
Safety Margins
Do the observed exposure levels remain below predefined preclinical toxicity thresholds?
Accumulation Predictability
Within the MAD phase, does the compound accumulate between repeated doses within the anticipated steady-state range, typically represented by an accumulation ratio between 1.3 and 2.5?
Within 48 to 72 hours of receiving bioanalytical concentration data, the PK team delivers a formal PK memorandum summarizing exposure parameters to the SRC. The committee reviews these findings alongside ECG results, vital signs, clinical laboratory assessments, and adverse event reports before approving progression to the next dose cohort.
Learn more about optimizing rapid developmental schedules via our Rapid Bioanalytical Method Development and Turnaround Services.
Harmonizing Bioanalytical Datasets with ICH M10 and FDA Guidelines
Global regulatory alignment of early-phase clinical datasets is achieved through strict adherence to the harmonized ICH M10 guideline. Compliance with these requirements ensures that pharmacokinetic datasets are immediately suitable for rolling New Drug Application (NDA) submissions.
The adoption of ICH M10 by major regulatory authorities, including the FDA and EMA, has standardized expectations for bioanalytical method validation and clinical sample analysis worldwide. This harmonized framework replaces older region-specific guidance and ensures that early-phase data can be accepted across global regulatory jurisdictions.
Key ICH M10 requirements evaluated during Phase I clinical support include:
Mid-Quality Control (MQC) Calibration
Unlike previous guidelines that permitted broader MQC definitions, ICH M10 requires MQC concentrations to be established between 30% and 50% of the upper limit of quantitation (ULOQ). This requirement ensures accurate and precise evaluation at concentration ranges representative of clinical steady-state exposure.
Reinjection Reproducibility
Validation must demonstrate that extracted samples remain stable and reproducible within the autosampler throughout the period between initial injection and potential reinjection. This requirement is particularly important when instrument failures or power interruptions occur during analytical runs.
Fixed-Dose Co-Medication Stability
For fixed-dose combinations or protocol-specified co-medications, ICH M10 requires stability studies, including freeze-thaw, bench-top, and long-term evaluations, to be conducted in matrices containing all co-administered compounds at clinically relevant concentrations such as their respective Cmax values. This assessment identifies potential interactions that may alter analyte stability during storage.
Incurred Sample Reanalysis (ISR)
ISR represents a mandatory quality control procedure during active clinical sample analysis. At least 10% of the first 1000 clinical samples and 5% of all subsequent samples must be randomly selected for reanalysis. The assay is considered reproducible when the difference between initial and repeat concentrations remains within ±20% for at least 67% of selected samples. Any ISR failure requires an immediate SOP-driven laboratory investigation covering sample handling, extraction procedures, and analytical performance.
Ensure full compliance for your next regulatory milestone by viewing our checklist on Data Integrity and Regulatory Audit Readiness in Bioanalytical Studies.
Conclusion
In conclusion, establishing high-performance Phase I Bioanalytical Support for Fast Track FDA programs represents a critical step in accelerating the development of central nervous system therapeutics. By proactively addressing the physical and chemical complexities associated with low-protein CSF matrices, including nonspecific adsorption and blood contamination, researchers can generate highly accurate and reproducible exposure data.
Implementing accelerated validation strategies under strict ICH M10 requirements ensures that clinical start-up activities remain efficient, compliant, and fully auditable. Additionally, combining high-throughput LC-MS/MS workflows with rapid in-house non-compartmental pharmacokinetic modeling enables clinical Safety Review Committees to make real-time, evidence-based dose-escalation decisions without delaying study progression.
Partner with an experienced development team by exploring our comprehensive GLP Bioanalytical Services for Regulatory Submissions.
Ultimately, this integrated and synchronized approach successfully bridges the gap between laboratory science and clinical application, facilitating the efficient delivery of breakthrough CNS therapies to patients who need them most.
For professional assistance in accelerating your clinical bioanalytical program under expedited regulatory pathways, contact the expert team at ResolveMass Laboratories Inc.
Please reach out via the ResolveMass Contact us page.
Frequently Asked Questions
Cerebrospinal fluid (CSF) is a limited biological matrix that is difficult and expensive to obtain in the large quantities required for bioanalytical method validation. To overcome this challenge, laboratories commonly use validated surrogate matrices that closely replicate the characteristics of human CSF. Formulations such as artificial CSF supplemented with Bovine Serum Albumin (BSA) help mimic physiological conditions while preserving valuable authentic CSF. This approach supports reliable assay development and validation without exhausting scarce clinical resources.
Nonspecific adsorption occurs when drug molecules adhere to surfaces such as collection tubes, storage vials, or analytical equipment instead of remaining in solution. This issue is particularly common with highly lipophilic compounds and can result in lower measured drug concentrations than are actually present. Consequently, important pharmacokinetic parameters such as Cmax and AUC may be underestimated. Inaccurate exposure data can influence dose-selection decisions and potentially affect the overall interpretation of clinical study outcomes.
Following lumbar puncture, small amounts of blood may inadvertently enter the CSF sample. If the sample is not processed promptly, red blood cells can rupture and release enzymes capable of degrading the drug analyte. Immediate centrifugation at controlled cold temperatures separates intact cells from the fluid portion of the sample before degradation begins. This process helps maintain sample integrity and ensures more accurate bioanalytical measurements throughout the study.
In Fast Track clinical programs, rapid data availability is essential for timely dose-escalation decisions. Bioanalytical laboratories generally process and analyze study samples within three to four days after receipt. Once concentration data are generated and quality checked, pharmacokinetic modeling and data interpretation are typically completed within an additional 48 to 72 hours. This accelerated workflow enables clinical teams to review exposure and safety data without delaying trial progression.
The ICH M10 guideline provides specific requirements for the placement of quality control samples within a bioanalytical method. According to the guideline, the Mid-Quality Control (MQC) level should be established between 30% and 50% of the Upper Limit of Quantitation (ULOQ). This defined range allows the assay to be evaluated at clinically relevant concentration levels. Maintaining consistency at this midpoint helps ensure reliable accuracy and precision across the calibration range.
Sentinel dosing is a precautionary strategy commonly used during first-in-human and early-phase clinical trials. Under this approach, a small number of participants, typically one receiving active treatment and one receiving placebo, are dosed before the remainder of the cohort. These individuals are closely monitored for any unexpected adverse effects or safety concerns. If no significant issues arise, the remaining participants can be dosed with greater confidence, enhancing overall participant safety.
Patients enrolled in clinical studies may receive additional medications alongside the investigational product. Because these compounds can potentially interact within biological samples, ICH M10 recommends evaluating analyte stability in the presence of relevant co-medications. This testing helps determine whether concurrent compounds influence chemical stability, recovery, or assay performance during storage and analysis. The objective is to ensure that reported concentrations accurately reflect the original sample composition.
Dilution integrity testing is performed to demonstrate that samples exceeding the calibration range can still be measured accurately after dilution. During validation, highly concentrated samples are prepared and then diluted using a blank biological matrix according to predetermined dilution factors. The diluted samples are subsequently analyzed and compared with expected concentrations. Acceptable dilution integrity is achieved when accuracy and precision remain within predefined validation criteria, confirming reliable quantification of high-concentration samples.
Reference:
- Lian, Z., Wang, N., Tian, Y., & Huang, L. (2021). Characterization of synthetic peptide therapeutics using liquid chromatography–mass spectrometry: Challenges, solutions, pitfalls, and future perspectives. Journal of the American Society for Mass Spectrometry, 32(8), 1852–1860. https://doi.org/10.1021/jasms.0c00479
- Maes, K., Smolders, I., Michotte, Y., & Van Eeckhaut, A. (2014). Strategies to reduce aspecific adsorption of peptides and proteins in liquid chromatography–mass spectrometry based bioanalyses: An overview. Journal of Chromatography A, 1358, 1–13. https://doi.org/10.1016/j.chroma.2014.06.072
- U.S. Food and Drug Administration. (2026). Bioanalytical method validation for biomarkers: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/179296/download
- U.S. Food and Drug Administration. (2026). Bioanalytical method validation for biomarkers: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/179296/download
- Shen, J., Swift, B., Mamelok, R., Pine, S., Sinclair, J., & Attar, M. (2019). Design and conduct considerations for first-in-human trials. Clinical and Translational Science, 12(1), 6–19. https://doi.org/10.1111/cts.12582
- Zhang, G., Zhang, N., Dong, L., Bai, N., & Cai, Y. (2023). Development and validation of an LC-MS/MS method for the quantitative determination of contezolid in human plasma and cerebrospinal fluid. Pharmaceuticals, 16(1), 32. https://doi.org/10.3390/ph16010032
- Ding, Y., Liao, Q., Zhang, Y., Wang, Y., Huang, J., Li, L., Li, J., & Chen, X. (2020). The central nervous system penetration ability of small molecules and their use in drug discovery for central nervous system diseases. Frontiers in Pharmacology, 10, 1626. https://doi.org/10.3389/fphar.2019.01626

