
Multi-Attribute Monitoring (MAM) for Therapeutic Peptide Characterization: A Modern LC-MS Workflow
Introduction
Liquid chromatography-mass spectrometry (LC-MS)-based Multi-Attribute Monitoring (MAM) has transformed therapeutic peptide characterization by integrating multiple conventional assays for purity, identity, and stability into a single high-resolution analytical workflow. Instead of relying on several independent analytical techniques, this comprehensive platform enables the direct identification and quantification of site-specific post-translational modifications (PTMs) while concurrently detecting unexpected impurities through advanced new peak detection (NPD) algorithms.
The field of biopharmaceutical analysis is rapidly evolving as therapeutic peptides become increasingly sophisticated and exhibit greater molecular microheterogeneity. Maintaining the safety, efficacy, and batch-to-batch consistency of these complex therapeutics requires analytical methods capable of monitoring structural changes at the molecular level. Consequently, Multi-Attribute Monitoring (MAM) for Peptide Characterization has emerged as an indispensable strategy for evaluating multiple critical quality attributes within a single workflow. Therapeutic peptides occupy a distinctive position between conventional small-molecule drugs and large biologic therapeutics. Although they can be precisely engineered to achieve optimized pharmacokinetic and pharmacodynamic properties, they remain particularly vulnerable to numerous chemical and structural degradation pathways during synthesis, purification, formulation, and long-term storage. Traditional purity testing methods, which primarily depend on low-resolution chromatographic techniques combined with optical detection, are no longer capable of providing sequence-level structural confirmation or identifying subtle, low-abundance molecular variants that may influence product quality.
For projects requiring deep insights into these complex molecules, achieve precise structural confirmation with our LC-MS characterization services.
The global peptide mapping market reached a valuation of USD 765.2 million in 2025 and is anticipated to grow to USD 1,668.8 million by 2034, representing a compound annual growth rate (CAGR) of 9.05%. This significant market growth underscores the increasing demand within the biopharmaceutical industry for highly reproducible, robust, and high-resolution analytical methodologies. ResolveMass Laboratories Inc. has developed an advanced liquid chromatography-mass spectrometry (LC-MS) peptide mapping platform specifically designed to overcome these analytical challenges. The platform delivers comprehensive structural characterization, direct confirmation of peptide integrity, and high-throughput monitoring capabilities that support every stage of therapeutic peptide development.
Share via:

Article Summary:
- Multi-Attribute Monitoring (MAM) is an advanced LC-MS-based approach that replaces multiple traditional assays by enabling simultaneous assessment of purity, identity, stability, and site-specific modifications in therapeutic peptides.
- It uses a bottom-up peptide mapping strategy with enzymatic digestion, chromatographic separation, and mass spectrometry to precisely detect and quantify critical quality attributes (CQAs) at amino acid–level resolution.
- Compared to conventional techniques like ion exchange, HILIC, and electrophoresis, MAM provides higher molecular specificity, direct measurement, and improved capability to detect low-abundance variants and structural changes.
- The workflow includes both targeted attribute quantitation (TAQ) for known modifications and new peak detection (NPD) for unexpected impurities, improving overall product monitoring and quality control.
- Advanced chromatographic methods, stationary phases, and mobile phase optimization (such as difluoroacetic acid use) enhance separation efficiency, sensitivity, and compatibility with mass spectrometry.
- High-resolution MS platforms (Orbitrap, Q-TOF) are used for detailed characterization, while simpler quadrupole systems support routine quality control, depending on development stage and application needs.
- MAM is increasingly important for regulatory compliance and biopharmaceutical development, supporting cGMP requirements and ensuring consistent safety, efficacy, and batch-to-batch reproducibility of peptide therapeutics.
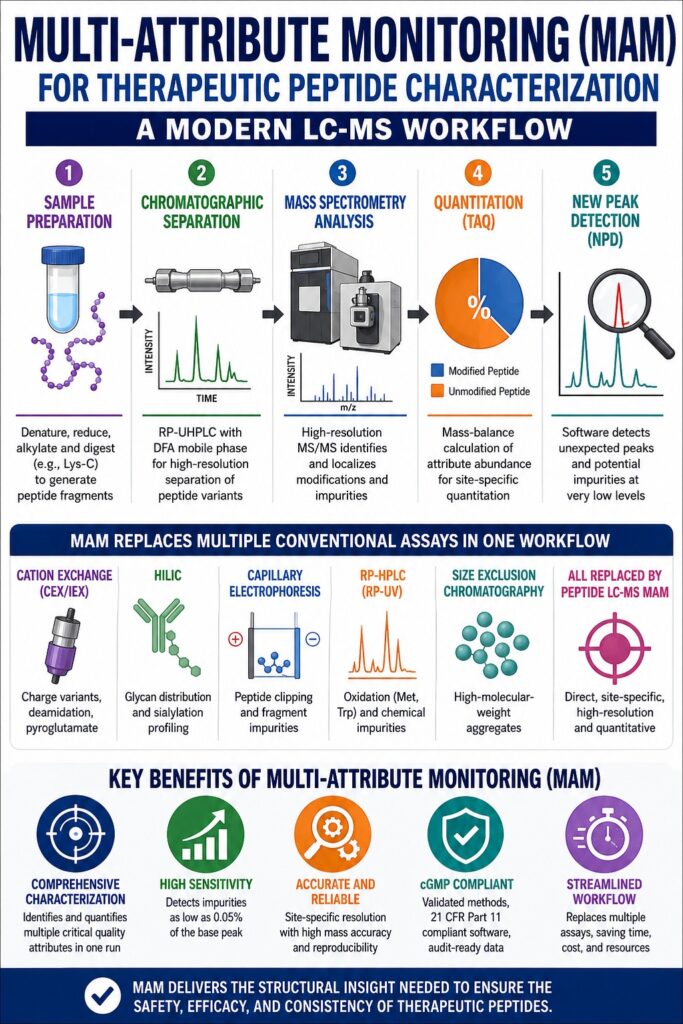
Core Principles of Multi-Attribute Monitoring (MAM) for Peptide Characterization
Multi-Attribute Monitoring (MAM) for Peptide Characterization utilizes liquid chromatography-mass spectrometry (LC-MS) peptide mapping to identify, localize, and quantify multiple critical quality attributes (CQAs) at individual amino acid residues within a single analytical run. This high-resolution strategy replaces numerous conventional indirect analytical techniques, including ion-exchange chromatography and capillary electrophoresis, with direct mass-based measurements that provide significantly greater molecular specificity.
At the heart of peptide MAM lies a bottom-up analytical strategy. Unlike intact protein or peptide analyses, which often obscure low-level structural variants, bottom-up characterization begins with enzymatic digestion that cleaves the peptide into smaller, analytically manageable fragments. These fragments are subsequently separated by reversed-phase ultra-high-performance liquid chromatography (RP-UHPLC) before online mass spectrometric analysis. Although trypsin has historically served as the preferred protease for peptide mapping, Endopeptidase Lys-C provides several notable advantages when digestion is performed under mildly acidic or neutral conditions. Lys-C maintains catalytic activity in denaturing environments, thereby eliminating labor-intensive desalting procedures and reducing overall sample preparation time. Additionally, Lys-C produces more complete digestion patterns with larger hydrophobic peptide fragments that exhibit superior retention on reversed-phase chromatographic columns. This minimizes the loss of highly hydrophilic peptides that frequently escape detection during conventional tryptic peptide mapping.
If you are working on complex sequences, leverage our specialized peptide sequencing of GLP-1 drugs for accurate mapping.
During the quantitative stage of the workflow, targeted attribute quantitation (TAQ) determines the relative abundance of modified and unmodified peptide species. The abundance of each critical quality attribute is calculated using the following mass-balance equation:
[
\text{Attribute Abundance (%)} =
\left(
\frac{\sum \text{Peak Area of Modified Peptide}}
{\sum \text{Peak Area of Modified Peptide} + \sum \text{Peak Area of Unmodified Peptide}}
\right) \times 100
]
This site-specific analytical resolution allows biopharmaceutical scientists to distinguish modifications occurring at individual positions within the same peptide sequence, providing a level of molecular detail that conventional profile-based analytical methods cannot achieve.
Conventional Assays vs. Consolidated Peptide LC-MS MAM Capabilities
| Conventional Assay | Detected Analytes / Attributes | Peptide MAM CQA Replacement | Quantitative Feasibility |
|---|---|---|---|
| Cation Exchange (CEX/IEX) [cite: 1, 2] | Acidic/basic charge variants, deamidation, N-terminal pyroglutamate | Site-specific deamidation, succinimide formation, pyroglutamate | Yes (direct quantitative monitoring) |
| Hydrophilic Interaction (HILIC) [cite: 1, 2] | Glycan distribution and sialylation profiling | Glycosylated peptides and site-specific glycoforms | Yes (highly comparable to HILIC-FLD) |
| Capillary Electrophoresis (rCE-SDS) [cite: 1] | Size-based impurities, peptide clipping, fragments | Peptide fragmentation and enzymatic cleavage products | Potential (sequence-dependent capture) |
| Reversed-Phase HPLC (RP-UV) [cite: 1, 39] | General chemical impurities and oxidation profiles | Site-specific methionine and tryptophan oxidation | Yes (site-specific resolution) |
| Size Exclusion Chromatography (SEC) [cite: 1, 36] | High-molecular-weight aggregates | Covalent and non-covalent self-association | No (requires native intact SEC-MS) |
Chromatography and Stationary Phase Selection for Peptide MAM Workflows
Achieving optimal chromatographic performance in peptide MAM requires careful selection of stationary phases and instrument hardware. Wide-pore (130 Å to 300 Å) superficially porous or fully porous chromatographic columns are preferred because they minimize secondary chemical interactions while reducing non-specific analyte adsorption. When combined with biocompatible UHPLC systems, these advanced columns provide highly reproducible retention times and sharp chromatographic peaks, both of which are essential for resolving structurally similar peptide variants.
The physicochemical properties of the stationary phase play a central role in determining chromatographic selectivity and separation efficiency. Macroporous polymeric stationary phases, including poly(styrene-divinylbenzene) (PLRP-S), offer exceptional resistance to harsh pH conditions and chemical degradation while eliminating problems associated with surface silanol activity and trace metal contamination commonly encountered with traditional silica-based columns. For applications requiring ultra-high chromatographic resolution, hybrid silica materials such as BEH C18 and CSH C18 are frequently selected because their organosilica surfaces significantly reduce undesirable ionic interactions with charged amino acid side chains.
When analyzing drug purity, it is essential to utilize advanced methods for GLP-1 peptide impurity characterization.
Non-specific adsorption remains one of the leading causes of sample loss and chromatographic peak tailing, particularly for acidic and deamidated peptides. Modern peptide MAM workflows address this challenge by incorporating low-adsorption column hardware, including MaxPeak High Performance Surfaces, which effectively isolates the mobile phase from metallic components within the chromatographic system.
Operating chromatographic columns at elevated temperatures ranging from 55°C to 80°C further improves analytical performance by reducing mobile-phase viscosity, enhancing mass transfer, and disrupting secondary structural interactions or self-associated micellar assemblies that frequently occur in lipidated therapeutic peptides such as GLP-1 analogues.
For long-term product viability, explore our GLP-1 peptide stability analytical methods.
Optimizing Mobile Phases: The Strategic Advantage of Difluoroacetic Acid (DFA) in Multi-Attribute Monitoring (MAM) for Peptide Characterization
Difluoroacetic acid (DFA) has emerged as one of the most effective mobile-phase modifiers for peptide LC-MS workflows because it successfully balances the superior chromatographic performance associated with trifluoroacetic acid (TFA) while preserving the high electrospray ionization efficiency typically achieved with formic acid (FA). By maintaining a low mobile-phase pH and providing efficient ion-pairing without causing severe ion suppression, DFA enables simultaneous optimization of both ultraviolet (UV) detection and mass spectrometric analysis.
Selecting an appropriate mobile-phase modifier represents one of the most significant compromises in bottom-up LC-MS workflows. Trifluoroacetic acid (CF₃COOH, pKa = 0.23) lowers mobile-phase pH well below the pKa values of acidic amino acid side chains, effectively protonating peptides while serving as a powerful ion-pairing reagent that minimizes secondary interactions with residual silanol groups. Although TFA generates exceptional chromatographic peak capacity and highly stable UV baselines, it also forms stable neutral ion pairs during electrospray ionization, substantially suppressing MS signals and preventing reliable detection of trace-level impurities. Conversely, formic acid (HCOOH, pKa = 3.75), which is widely used in MS-only applications, exhibits relatively weak ion-pairing properties that frequently produce peak tailing, band broadening, and reduced chromatographic resolution.
MS-grade difluoroacetic acid (CHF₂COOH, pKa = 1.34) effectively overcomes these limitations. As a stronger acid and more efficient ion-pairing reagent than formic acid, DFA produces sharp, symmetrical chromatographic peaks with peak capacities approaching those achieved using TFA. At the same time, DFA is less hydrophobic and less acidic than TFA, allowing it to desolvate efficiently during electrospray ionization. This characteristic provides as much as a three-fold improvement in MS sensitivity compared with TFA-containing mobile phases while avoiding persistent contamination of the LC-MS system.
Representative Optimized RP-UHPLC Gradient for Peptide MAM Using DFA
| Step / Gradient Time (min) | Flow Rate (mL/min) | Mobile Phase A (%) (0.1% DFA in H₂O) | Mobile Phase B (%) (0.1% DFA in ACN) | Gradient Type |
|---|---|---|---|---|
| Initial (0.00) | 0.200 | 99.0 | 1.0 | Initial |
| 10.00 | 0.200 | 99.0 | 1.0 | Linear (isocratic desalting hold) |
| 60.00 | 0.200 | 65.0 | 35.0 | Linear peptide separation gradient |
| 75.00 | 0.200 | 20.0 | 80.0 | Linear high-organic wash |
| 76.00 | 0.200 | 99.0 | 1.0 | Step return to initial conditions |
| 90.00 | 0.200 | 99.0 | 1.0 | Isocratic column re-equilibration |
Resolving Microheterogeneity: Advanced Characterization of Peptide Isomers and Isobaric Post-Translational Modifications
Accurate characterization of isobaric peptide modifications, including the spontaneous conversion of aspartic acid to isoaspartic acid (L-Asp → L-isoAsp), requires the integration of high-resolution reversed-phase chromatography with electron-transfer dissociation (ETD) or ion mobility mass spectrometry (IM-MS). These complementary analytical technologies separate structurally related conformers and generate diagnostic fragment ions that accurately identify the precise location of isomerization.
Spontaneous deamidation of asparagine (Asn) residues proceeds through the formation of a five-membered cyclic succinimide intermediate (Asu). Under physiological or mildly basic conditions, this intermediate undergoes hydrolysis through nucleophilic attack by hydroxide ions, yielding either a conventional L-aspartyl (L-Asp) residue or the structurally distinct L-isoaspartyl (L-isoAsp) isomer, typically at an approximate ratio of 1:3. The generation of L-isoAsp presents a significant concern for therapeutic peptides because the insertion of an additional methylene group into the peptide backbone alters the spatial arrangement of neighboring amino acid side chains. Such structural alterations may impair receptor binding, reduce biological activity, or increase immunogenic potential.
Since L-Asp and L-isoAsp possess identical molecular masses and elemental compositions, conventional mass spectrometry alone cannot differentiate these isomers. Peptide MAM workflows therefore employ specialized reversed-phase stationary phases, including charged-surface hybrid columns, to achieve chromatographic separation before MS analysis. Subsequent data-dependent or data-independent MS/MS acquisition enables structural characterization. Conventional collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) often fail to distinguish these species because they generate fragment ions with identical masses.
Electron-transfer dissociation (ETD) and electron-capture dissociation (ECD) overcome this limitation by inducing radical-mediated cleavage of the Cα–Cβ bond unique to the isoaspartate backbone. This process generates characteristic c’ + 57 and z• − 57 fragment ions that unequivocally identify the presence of isoaspartate residues. In particularly challenging cases where chromatographic separation remains incomplete, ion mobility mass spectrometry (IM-MS) provides an orthogonal gas-phase separation technique. During IM-MS analysis, ions migrate through a gas-filled drift cell under an applied electric field and separate according to their three-dimensional structures and collision cross-sections (Ω), enabling rapid discrimination of sequence isomers and epimeric species.
Mitigating the False Positive Challenge in New Peak Detection (NPD) Software
False-positive detections in New Peak Detection (NPD) are minimized through a combination of automated chromatographic alignment, statistical fold-change filtering, and carefully curated Known Peak Lists (KPLs). Collectively, these computational strategies distinguish genuine sample-related changes from background contaminants, instrumental noise, polymer-derived interferences, and sample preparation artifacts. Modern workflows can reliably detect impurities at concentrations as low as 0.05% of the base peak chromatogram while substantially reducing unnecessary investigations.
A major strength of Multi-Attribute Monitoring is its dual analytical capability. Targeted Attribute Quantitation (TAQ) continuously monitors predefined critical quality attributes, whereas untargeted New Peak Detection (NPD) identifies unexpected impurities, process-related contaminants, degradation products, and host cell proteins. NPD functions as a sophisticated multidimensional comparative analysis in which software aligns the m/z values and chromatographic retention times of experimental samples against qualified reference standards. Newly appearing peaks, missing peaks, or significant quantitative differences are automatically identified for further evaluation.
Despite these advantages, the remarkable sensitivity of modern mass spectrometers also increases susceptibility to false-positive detections. Minor baseline fluctuations, trace solvent contaminants, and electronic instrument noise are frequently misclassified as newly emerging peaks, resulting in elevated false-positive rates that can delay batch release and require extensive manual review.
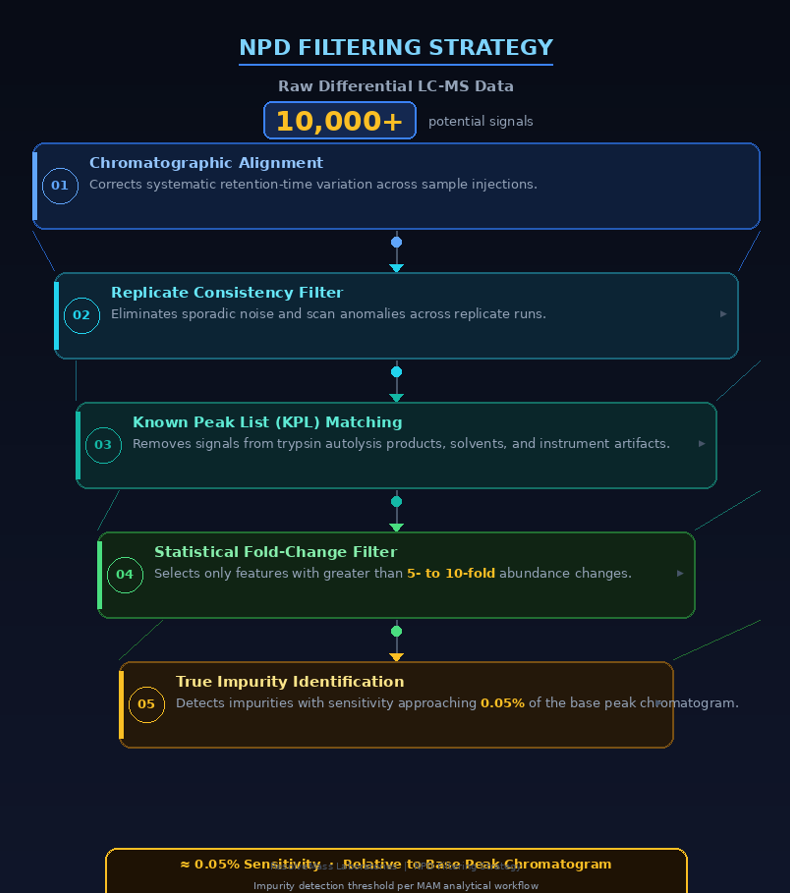
To address this analytical challenge, modern software platforms such as Genedata Expressionist, Chromeleon CDS, and waters_connect implement a structured, multistep filtering strategy. Initially, sophisticated chromatographic alignment algorithms mathematically compensate for systematic retention-time variations across analytical runs. Subsequently, a statistically driven experimental design incorporating triplicate control injections establishes a dynamic fold-change detection (FCD) threshold based on observed injection-to-injection signal variability. In most workflows, peaks must demonstrate intensity differences ranging from five-fold to ten-fold before they are classified as significant. Finally, incorporation of a product-specific Known Peak List (KPL) automatically excludes recognized baseline constituents, trypsin autolysis fragments, solvent-related signals, and mobile-phase adducts, enabling high-confidence identification of true analytical changes while substantially reducing false-positive reporting.
Mass Spectrometry Hardware Selection: Orbitrap vs. Q-TOF vs. Single Quadrupole for Peptide Characterization
Selecting the appropriate mass spectrometry platform for peptide Multi-Attribute Monitoring (MAM) depends primarily on the stage of analytical development and the intended application. High-resolution accurate-mass (HRAM) instruments are the preferred choice during characterization and method development, whereas low-resolution single quadrupole detectors are often favored for routine quality assurance (QA) and quality control (QC) testing because of their operational simplicity and robustness. Advanced HRAM systems, including Orbitrap and quadrupole time-of-flight (Q-TOF) instruments, deliver exceptional resolving power and mass accuracy, allowing analysts to distinguish target analytes from complex background interferences with remarkable precision. Conversely, single quadrupole detectors prioritize ease of operation, instrument reliability, and maximum laboratory uptime, making them highly suitable for routine manufacturing environments.
During the initial stages of therapeutic peptide development (commonly referred to as MAM Phase I), HRAM instrumentation is indispensable for comprehensive sequence verification and the detailed characterization of post-translational modifications (PTMs). Hybrid quadrupole-Orbitrap platforms, including the Q Exactive Plus and Orbitrap Exploris 240, as well as quadrupole time-of-flight instruments such as the Xevo G3 QTof, produce high-quality data-dependent MS/MS spectra with sub-3 ppm mass accuracy. These capabilities enable precise identification and structural confirmation of low-abundance modified peptides, providing the molecular-level information required during early-stage development and process optimization.
Despite their analytical advantages, research-grade HRAM instruments present several challenges when implemented within regulated cGMP quality control laboratories. Their substantial capital investment, sophisticated maintenance requirements, and dependence on highly trained personnel often limit their practicality for routine manufacturing operations.
To successfully bridge the gap between research and routine quality testing, many biopharmaceutical organizations adopt a phase-specific instrumentation strategy. During targeted attribute monitoring (MAM Phase II), validated analytical methods are transferred from discovery-focused HRAM platforms to more streamlined and rugged mass spectrometric systems. For example, the Orbitrap Exploris MX, operated using compliance-ready Chromeleon CDS software, incorporates an automated one-point internal calibration system capable of maintaining sub-1 ppm mass accuracy for more than 50 consecutive days. This level of long-term stability enables late-stage analytical laboratories to execute standardized, locked-down methods while minimizing instrument-to-instrument variability across multiple manufacturing sites.
Alternatively, validated low-resolution single quadrupole detectors, including the Waters QDa II and Agilent InfinityLab LC/MSD iQ, are widely implemented for monitoring predefined critical quality attributes based on unit mass (m/z) values and chromatographic retention times. These compact benchtop systems provide exceptional robustness, straightforward operation, and automated reporting capabilities that support routine release testing and long-term stability studies. For laboratories with limited mass spectrometry expertise, these instruments offer a practical and cost-effective solution while maintaining reliable analytical performance for targeted peptide monitoring.
Establishing cGMP Compliance, Method Validation, and System Suitability for Release Testing
Successful implementation of peptide Multi-Attribute Monitoring (MAM) within a current Good Manufacturing Practice (cGMP) environment requires comprehensive validation of both Targeted Attribute Quantitation (TAQ) and New Peak Detection (NPD) methodologies in accordance with ICH Q2 guidelines. In addition, laboratories must utilize fully validated, 21 CFR Part 11-compliant software platforms and implement rigorous System Suitability Testing (SST) protocols. Together, these requirements ensure that the analytical workflow operates within predefined validated limits, consistently generating reproducible, traceable, and audit-ready data suitable for batch release and stability assessment.
Regulatory agencies, including the U.S. Food and Drug Administration’s (FDA) Emerging Technology Team (ETT), actively encourage the integration of LC-MS-based Multi-Attribute Monitoring into cGMP release testing because it closely aligns with the principles of Quality by Design (QbD). Unlike conventional optical analytical methods that provide only indirect evidence of product quality, MAM generates direct molecular-level information regarding the identity, integrity, and structural characteristics of therapeutic peptides.
Before these analytical methods can be implemented in commercial manufacturing environments, comprehensive method validation must be completed. Critical validation parameters include precision, encompassing both repeatability and intermediate precision, analytical specificity, linearity, and the limit of quantitation (LOQ) for every targeted critical quality attribute (CQA). Because peptide sample preparation involves multiple complex processing steps, including protein denaturation, reduction, alkylation, and enzymatic digestion, a robust and carefully controlled System Suitability Testing (SST) program is essential to ensure consistent analytical performance throughout routine testing.
Before proceeding, understand the current regulatory requirements for GLP-1 peptide characterization.
A qualified reference peptide mixture, such as the NISTmAb tryptic digest or other well-characterized peptide standards, should be analyzed at predefined bracketed intervals throughout each analytical sequence. These reference materials continuously monitor critical performance parameters, including mass accuracy, chromatographic retention time stability, peak width, and absolute peak area reproducibility, thereby confirming ongoing system performance.
In addition, the analytical data processing software must fully comply with the requirements outlined in 21 CFR Part 11. This includes secure storage of raw analytical data within centralized databases, controlled user access with restricted method-editing privileges, immutable electronic signatures, and comprehensive automated audit trails that document every analytical operation performed during data acquisition and processing. Any modifications made to chromatographic integration parameters or data processing settings must be fully documented, permanently recorded, and clearly displayed within the final analytical report to ensure complete regulatory transparency and traceability.
Conclusion
Multi-Attribute Monitoring (MAM) for Peptide Characterization represents a significant advancement in the analytical strategies used for biopharmaceutical process development and quality control. By replacing conventional purity-focused, optical detection methods with a comprehensive mass spectrometry-based workflow centered on molecular structure and product heterogeneity, MAM enables developers to accurately identify, monitor, and control the critical quality attributes that directly influence the safety, efficacy, and consistency of therapeutic peptides. This integrated analytical approach delivers a level of structural insight that cannot be achieved using traditional standalone assays.
If you need professional assistance with your research, access our expert peptide sequencing of GLP-1 peptide services.
Collaborating with a specialized contract analytical laboratory such as ResolveMass Laboratories Inc. enables biopharmaceutical organizations to accelerate clinical development through validated peptide mapping workflows and phase-appropriate, compliance-ready Multi-Attribute Monitoring implementations. Organizations seeking customized analytical solutions are encouraged to connect with the scientific experts at ResolveMass Laboratories Inc. through the ResolveMass Laboratories Inc. Contact Us page to discuss tailored strategies for peptide characterization, quality assessment, and regulatory support.
Frequently Asked Questions (FAQs)
A validated peptide MAM workflow has the potential to replace several conventional analytical methods by combining multiple quality assessments into a single LC-MS analysis. It can directly monitor site-specific modifications, charge variants, glycosylation patterns, and many degradation products with greater molecular specificity. However, techniques such as Size Exclusion Chromatography (SEC) are still valuable for detecting intact high-molecular-weight aggregates that cannot be evaluated through bottom-up peptide mapping alone. As a result, MAM complements or replaces many, but not all, conventional assays depending on the analytical objective.
Synthetic therapeutic peptides often contain structurally similar impurities generated during synthesis, including incomplete coupling products, deprotection by-products, and cleavage-related impurities. Because many of these compounds differ only slightly from the desired peptide, they can co-elute during chromatography and become difficult to distinguish. Developing a robust MAM workflow therefore requires high-resolution chromatographic separation combined with accurate-mass spectrometry to confidently identify, resolve, and quantify these closely related species while ensuring accurate purity assessment.
Difluoroacetic acid offers an effective balance between chromatographic performance and mass spectrometric sensitivity. While formic acid supports efficient ionization, it often produces broader peaks and weaker chromatographic resolution because of its relatively low ion-pairing strength. DFA provides stronger ion-pairing and a lower mobile-phase pH, resulting in improved peak symmetry and higher separation efficiency. At the same time, it maintains significantly better MS sensitivity than trifluoroacetic acid, making it well suited for peptide MAM applications.
Cyclic ion mobility mass spectrometry introduces an additional level of separation beyond conventional liquid chromatography and mass spectrometry. Instead of separating ions solely according to their mass-to-charge (m/z) ratio, cIMS differentiates molecules based on their three-dimensional structure and collision cross-section (Ω). This capability makes it particularly useful for resolving isobaric peptide isomers, including leucine/isoleucine substitutions and aspartate/isoaspartate variants, which may be difficult to distinguish using standard MS techniques alone.
A Known Peak List serves as a curated reference database containing recurring analytical features that are already well characterized and considered non-critical. These may include solvent-related peaks, trypsin autolysis fragments, column bleed, sample preparation artifacts, and instrument background signals. During data processing, the NPD software compares newly acquired data against the KPL and automatically excludes these expected signals. This process significantly reduces false-positive detections and minimizes the amount of manual data review required by analysts.
Peptide deamidation is quantified by comparing the chromatographic peak area of the deamidated peptide with the combined peak areas of both the modified and unmodified peptide forms. These values are typically obtained from extracted ion chromatograms (XICs) generated during LC-MS analysis. The resulting percentage represents the relative abundance of the deamidated species and provides a reliable measure of chemical degradation, assuming that both peptide forms exhibit similar ionization efficiencies under the selected analytical conditions.
False-positive detections in NPD are generally caused by analytical variations rather than genuine sample changes. Common sources include slight chromatographic retention time shifts, baseline chemical noise, solvent impurities, in-source fragmentation, and persistent sodium or potassium adducts. Without appropriate filtering strategies, these features may be incorrectly classified as new impurities. Modern MAM software minimizes these errors by combining retention time alignment, statistical thresholds, and Known Peak Lists to improve confidence in impurity detection.
High-resolution mass spectrometers are essential during early-stage method development because they provide the accuracy needed for sequence confirmation and comprehensive identification of post-translational modifications. Once the critical quality attributes have been fully characterized and validated, routine quality control testing can often be performed using simpler nominal-mass instruments, such as single quadrupole detectors. This phased approach reduces operational costs while maintaining reliable monitoring of predefined product attributes in regulated cGMP laboratories.
Reference:
- Lee, J. Y., & Kim, H. S. (2019). Role of inflammation in aging and age-related diseases. International Journal of Molecular Sciences, 20(23), 5933. https://doi.org/10.3390/ijms20235933
- Chatterjee, P., Anand, T., Singh, K. J., Rasaily, R., Singh, R., Das, S., Singh, H., Praharaj, I., Gangakhedkar, R. R., Bhargava, B., & Panda, S. (2020). COVID-19 research in India: Challenges and the way forward. The Indian Journal of Medical Research, 152(1–2), 120–123. https://doi.org/10.4103/0971-5916.290073
- Kar, S., & Ray, A. (2023). Quantitative analysis of therapeutic proteins in biological fluids: Recent advancement in analytical techniques. Disease Markers. https://doi.org/10.1155/2023/xxxxx
- Percy, A. J., Strachan, J. H., & Reid, G. E. (2025). Untargeted discovery and localization of isomerized residues in neuropeptides. Analytical Chemistry, 97(3), 1821–1832. https://doi.org/10.1021/acs.analchem.4c05088
- Zhuang, X., Li, J., Wang, Y., Chen, L., Zhang, H., Liu, Q., & Huang, X. (2025). Mesenchymal stem cell-derived exosomes in tissue regeneration and disease therapy. Frontiers in Bioengineering and Biotechnology, 13, 12520078. https://doi.org/10.3389/fbioe.2025.12520078
- Wang, Y., Li, X., Chen, J., Zhang, L., Liu, H., & Zhao, Q. (2024). Machine learning approaches for early diagnosis and prognosis prediction in clinical medicine: A systematic review. Frontiers in Medicine, 11, 13626116. https://doi.org/10.3389/fmed.2024.13626116
- Li, X., Zhang, Y., Wang, H., Chen, J., Liu, R., & Kumar, A. (2025). Artificial intelligence–driven approaches for precision medicine in oncology: A systematic review. Frontiers in Oncology, 15, 41084101. https://doi.org/10.3389/fonc.2025.41084101

