
Introduction
Sponsors of generic drugs can improve their time-to-market and increase the likelihood of regulatory approval by utilizing comprehensive CDMO Services for Generic Oral Solid Dosage Forms to manage the complex and interconnected pathway involving formulation development, analytical characterization, and bioequivalence verification. Choosing an experienced contract development and manufacturing organization (CDMO) is a crucial decision because converting a reference listed drug (RLD) into a pharmaceutically and therapeutically equivalent generic product requires the integration of advanced material science expertise with stringent regulatory requirements. As outsourcing continues to expand—with 73% of FDA-approved drugs in 2025 reportedly utilizing contract manufacturing organizations for their active pharmaceutical ingredient (API) and chemistry, manufacturing, and controls (CMC) documentation—the selection of a scientifically capable development partner has become a fundamental requirement for commercial success. A proactive CDMO that performs early Drug Master File (DMF) evaluations, maintains a strong regulatory compliance history with zero FDA Form 483 observations, and generates submission-ready data can significantly reduce the likelihood of receiving complete response letters (CRLs).
To understand the critical operational differences in these partnerships, explore how a specialized CDMO vs CRO for generic drug development can impact your project trajectory.
ResolveMass Laboratories Inc. serves as a highly specialized and scientifically authoritative laboratory partner with advanced chromatographic, mass spectrometric, and dissolution profiling capabilities required to support these complex product categories. By combining systematic Quality by Design (QbD) principles with phase-appropriate analytical development, developers can proactively address risks related to raw material variability, process scale-up, and clinical bioequivalence trials. This technical report examines the advanced engineering principles, biopharmaceutic considerations, and regulatory validation pathways necessary for the successful execution of Abbreviated New Drug Application (ANDA) programs involving tablets, capsules, and complex modified-release oral solid dosage (OSD) systems.
Ensure your submission meets the highest standards by leveraging expert regulatory support for generic drugs in the US and Canada.
Share via:
Core Engineering Principles of Generic Tablets and Capsules
Drug developers optimize the design and manufacturability of generic tablets and capsules by systematically aligning API material attributes with the most suitable compaction process. The primary manufacturing approaches include wet granulation, dry granulation (roller compaction), and direct compression. The selection of each method depends on important physical characteristics, including powder flowability, compactability, and thermal stability.
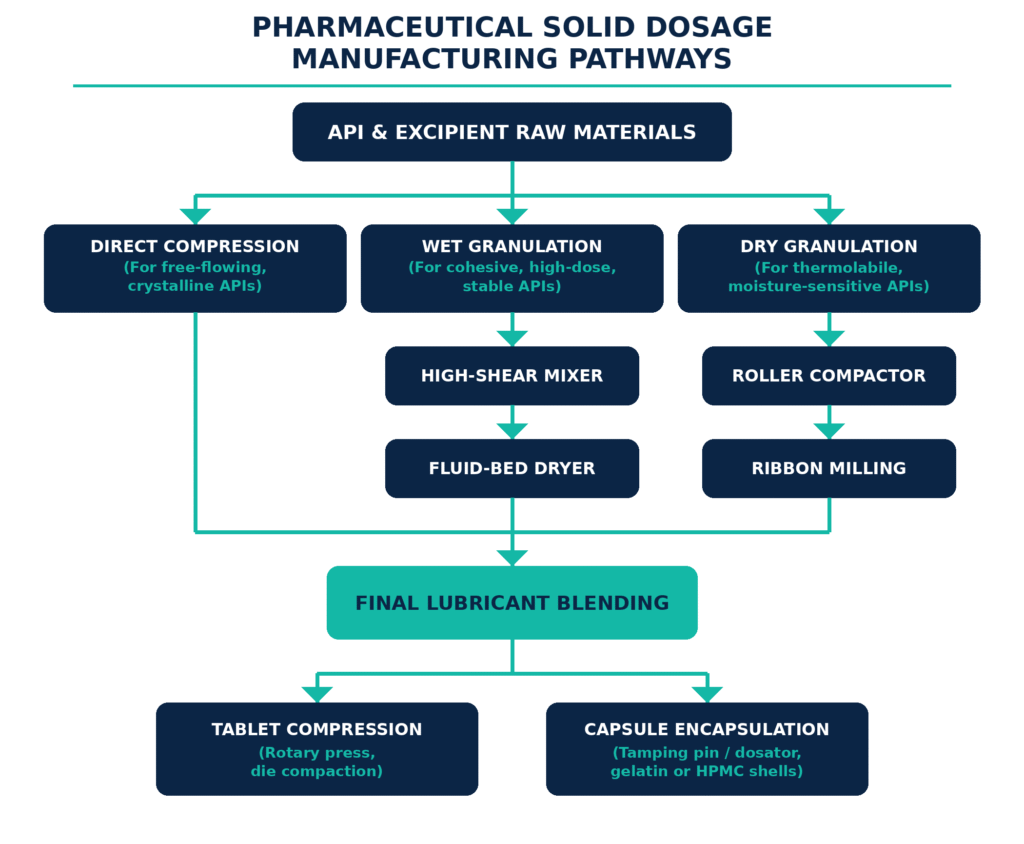
The appropriate processing pathway is established through a comprehensive evaluation of the API’s bulk properties. High-dose APIs with highly cohesive, electrostatic, or poor-flowing characteristics are generally unsuitable for direct compression because these properties can result in mass variation, flow segregation, and inconsistent tablet weight profiles during operation on high-speed rotary presses. In such cases, wet granulation is employed. During this process, the API and diluents are wetted with a binder solution while undergoing high-shear mixing, causing the fine particles to consolidate into dense, spherical granules. The resulting wet mass is dried in fluidized bed dryers to achieve a predetermined loss-on-drying (LOD) percentage. The dried material is then milled and lubricated to promote uniform flow and reliable die filling during tablet compression.
For projects requiring specialized scaling, consult with a pharmaceutical CDMO in the US and Canada to streamline your manufacturing process.
For APIs that are susceptible to degradation caused by moisture or heat, dry granulation through roller compaction provides a suitable alternative. In this approach, the powder blend passes between two counter-rotating rollers, where it is compressed into a dense ribbon. The ribbon is then immediately milled through oscillating screens to produce free-flowing granules with strong compactability. The tablet compression stage itself may require advanced compaction simulators to develop robust compression profiles and define the relationship between compaction force, tablet tensile strength, and disintegration time.
| Compression/Filling Technology | Material Selection Criteria | Processing Key Equipment | Critical Quality Attributes (CQAs) Managed |
|---|---|---|---|
| Direct Compression | Free-flowing, crystalline, compactable APIs; typically low-to-moderate doses. | High-speed V-blenders, rotary tablet presses with force-feeder assemblies. | Blend uniformity, weight variation, hardness, friability, disintegration. |
| Wet Granulation | Cohesive, poorly flowing, high-dose, heat-stable, and moisture-stable APIs. | High-shear granulators, fluid-bed dryers, cone mills. | Granule size distribution, moisture content (LOD), bulk density, flow index. |
| Dry Granulation | Moisture-sensitive, thermolabile, poorly compactable, low-density APIs. | Roller compactors, ribbon granulators, rotary sizers. | Ribbon density, granule compressibility, fraction of fines, dissolution rate. |
| Capsule Encapsulation | Powders, granules, coated pellets, or minitablets. | Tamping pin or dosator capsule-filling machines. | Fill weight, shell integrity, lock-length, gastric disintegration time. |
Accelerate your path to commercialization by partnering with a CDMO to accelerate generic drug development in the US and Canada.
Overcoming Physicochemical and Process Constraints in Capsule Filling
CDMOs address capsule-filling challenges, including powder cohesiveness, electrostatic accumulation, and poor flowability, through phase-appropriate excipient optimization and tightly controlled manufacturing environments. These strategies help prevent critical manufacturing problems such as powder bridging, inconsistent die filling, and variation in capsule weight.
When dealing with complex analytical hurdles, access precision through peptide analytical characterization services
or learn from our generic peptide drug analytical characterization case study.
Capsule development and manufacturing are governed by strict physical, chemical, and toxicological considerations:
Physical Cohesion and Bridging: Low-dose APIs and highly cohesive powder blends can frequently form bridges across filling hoppers, interfering with consistent dosing in tamping pin or dosator systems. This issue can be addressed by optimizing the particle size distribution of the diluent, such as through the use of spray-dried lactose or silicified microcrystalline cellulose, and by adding glidants, including colloidal silicon dioxide, to decrease inter-particle friction.
Electrostatic Accumulation: Manufacturing under low-humidity conditions, which may be necessary for moisture-sensitive APIs, can increase the accumulation of static electricity. This can negatively affect powder flow and reduce fill-weight accuracy. CDMOs manage this risk by maintaining relative humidity within a carefully controlled operating range, typically 30% to 45% RH, and by incorporating anti-static processing equipment into the manufacturing process.
Shell-Formulation Interactions: Gelatin shells naturally contain approximately 13% to 16% moisture and can therefore function as moisture donor-acceptor systems. Hygroscopic formulations may draw moisture from the shell, potentially causing brittleness and cracking. Conversely, moisture-sensitive APIs may undergo degradation as a result of absorbing water from the gelatin shell. Hydroxypropyl methylcellulose (HPMC) shells, which generally contain only 3% to 6% moisture, may therefore be selected to reduce these chemical and physical interactions.
Bioavailability and Solubility Barriers: For poorly soluble, beyond-rule-of-5 molecules, formulation scientists may employ supersaturated delivery systems. Using the “spring and parachute” concept, high-energy amorphous solid-state forms serve as the “spring” by rapidly increasing dissolved drug concentrations within the intestinal lumen. These systems are combined with precipitation-inhibiting polymers, such as HPMC or copovidone, which function as the “parachute” by sustaining supersaturation and maximizing systemic flux.
Need expert help in the peptide space? Explore our peptide CDMO services
or discover why we are recognized as the best peptide CDMO for your specific needs.
Modified-Release Formulation Platforms and Dissolution Kinetics
Oral modified-release (MR) dosage forms use polymeric barrier systems to control the rate, location, or timing of drug release throughout the gastrointestinal tract. These technologies include extended-release (ER) hydrophilic or eroding matrices, delayed-release (DR) enteric coatings, and multi-particulate pellet systems developed to achieve the target product profile.
Hydrophilic matrix systems are widely used for once-daily or twice-daily oral administration. When exposed to gastric fluid, polymers such as hypromellose (HPMC) rapidly hydrate and form a dense gel layer around the tablet core. The release of the active ingredient is subsequently controlled by two concurrent processes described by Fick’s laws of diffusion:
J = −D dC/dx
The diffusion process can be described by Fick’s first law of diffusion: J = −D(dC/dx), where J represents the diffusion flux, D is the diffusion coefficient of the active pharmaceutical ingredient (API) through the hydrated gel barrier, and dC/dx represents the concentration gradient across the diffusion pathway. As the gel layer expands, water penetrates deeper into the tablet core and dissolves the API, allowing the dissolved drug molecules to diffuse outward through the hydrated polymer barrier. Simultaneously, the outer regions of the gel matrix undergo polymer chain disentanglement, followed by gradual erosion, which contributes to the overall drug release mechanism.
For highly soluble APIs, hydrophobic polymers, such as ethylcellulose, or lipids, such as hydrogenated castor oil, can be incorporated to create non-eroding matrix networks. In these systems, the drug gradually dissolves and is released primarily through diffusion across a tortuous network of microscopic pores. Delayed-release formulations, which are designed to protect acid-labile drugs or reduce exposure of the gastric mucosa, depend on enteric coatings containing anionic methacrylic acid-methyl methacrylate copolymers. These polymers contain carboxyl groups that remain un-ionized under the acidic conditions of the stomach but become ionized and dissolve rapidly when the formulation reaches pH thresholds of 5.5, 6.0, or 6.8 in the small intestine.
Multi-particulate pellets contained within capsules provide another advanced formulation strategy. By coating individual beads with different thicknesses of extended-release or delayed-release polymers, developers can combine several release profiles, including biphasic or pulsatile release, within a single capsule. This approach enables the delivery of complex pharmacokinetic profiles while minimizing the potential for localized gastrointestinal irritation.
Understand the strategic differences between providers by reviewing our peptide CDMO vs CRO comparison guide.
Critical Evaluation of Alcohol-Induced Dose Dumping (AIDD) Risks
Alcohol-Induced Dose Dumping (AIDD) refers to the rapid and premature release of a controlled-release drug dose in the stomach following co-administration with ethanol, potentially resulting in severe systemic toxicity. To reduce this risk, formulation scientists select polymers capable of maintaining their structural integrity and resisting dissolution when exposed to hydro-alcoholic mixtures containing up to 40% v/v ethanol.
The physiological implications of alcohol-induced dose dumping (AIDD) gained substantial clinical and regulatory attention after the withdrawal of the modified-release hydromorphone product Palladone in 2005. When the product was co-administered with a 40% alcoholic beverage, the peak plasma concentration (Cmax) increased approximately six-fold, or 600%, compared with administration alongside water. In a well-designed controlled-release formulation, drug release is expected to follow zero-order kinetics, meaning that the drug is released at a relatively constant rate over time:
dC/dt = k₀
where the release rate remains constant over time and is independent of the drug concentration. However, when susceptible polymers are exposed to ethanol, they may dissolve or lose their gel strength. This can cause the controlled-release mechanism to fail and the release profile to shift toward highly accelerated first-order kinetics:
dC/dt = −k₁C
Under these conditions, the entire dose may be released rapidly into the stomach within 30 minutes.
During formulation development, regulatory agencies require extensive in vitro comparative dissolution testing of test and reference products under different alcohol concentrations to assess this risk. Testing is generally performed using USP Apparatus 1 (baskets) at 100 rpm or Apparatus 2 (paddles) at 50/75 rpm in 900 mL of 0.1 N HCl to simulate gastric conditions. The dissolution medium is progressively supplemented with 5%, 20%, and 40% v/v ethanol, and testing is typically conducted for 2 hours.
A generic formulation is considered robust and resistant to alcohol-induced dose dumping (AIDD) when its dissolution profile in alcoholic media remains comparable to that observed in acidic media without alcohol. This similarity is commonly evaluated using the similarity factor (f₂), where an f₂ value greater than 50 generally indicates that the two dissolution profiles are comparable. Conversely, an f₂ value below 50 may suggest a meaningful difference in dissolution behavior and could indicate potential susceptibility to dose dumping. In such cases, reformulation with alcohol-resistant polymers, including selected grades of sodium alginate, carbomers, or high-molecular-weight polyethylene oxide, may be considered. These polymers are chosen for their ability to maintain swelling and gel-forming properties when exposed to alcoholic media.
Discover how specialized manufacturing infrastructure supports complex programs by exploring our hubs for a peptide CDMO in United States.
| Parameter | United States (USFDA) Requirements | European Union (EMA) Requirements | Health Canada Requirements |
|---|---|---|---|
| Scope of Active Ingredients | Mandated for all modified-release solid oral dosage forms. | Required for all modified-release oral formulations. | Mandated for opioids and critical MR dosage forms. |
| Testing Media | 0.1 N HCl (pH 1.2) | Simulated Gastric Fluid (SGF) | Optimized acidic and buffer media. |
| Ethanol Concentrations | 0%, 5%, 20%, and 40% v/v | 5% and 20% v/v | 0%, 5%, 20%, and 40% v/v. |
| Testing Duration | 2 Hours (samples taken every 15 mins) | Expected in vivo release profile duration (up to 12 hours). | 2 Hours or product-specific duration. |
| Evaluation Metrics | Dissolution profile similarity ($f_2 \ge 50$); clinical in vivo BE if in vitro tests fail. | Strict risk-benefit assessment; reformulation is required if interaction occurs. | Profile comparison and complete documentation confirming the absence of AIDD. |
Navigating FDA and ICH Bioequivalence (BE) Guidelines for ANDA Programs
Demonstrating bioequivalence for generic modified-release formulations requires evidence that the test product produces a comparable rate and extent of systemic drug absorption to the Reference Listed Drug (RLD). Bioequivalence is generally established when the 90% confidence interval for the geometric mean ratio between the test and reference products falls entirely within the accepted range of 80% to 125% for both Cmax and AUC. Here, Cmax represents the maximum observed plasma drug concentration, while AUC represents the area under the plasma concentration–time curve and reflects the overall extent of systemic exposure.
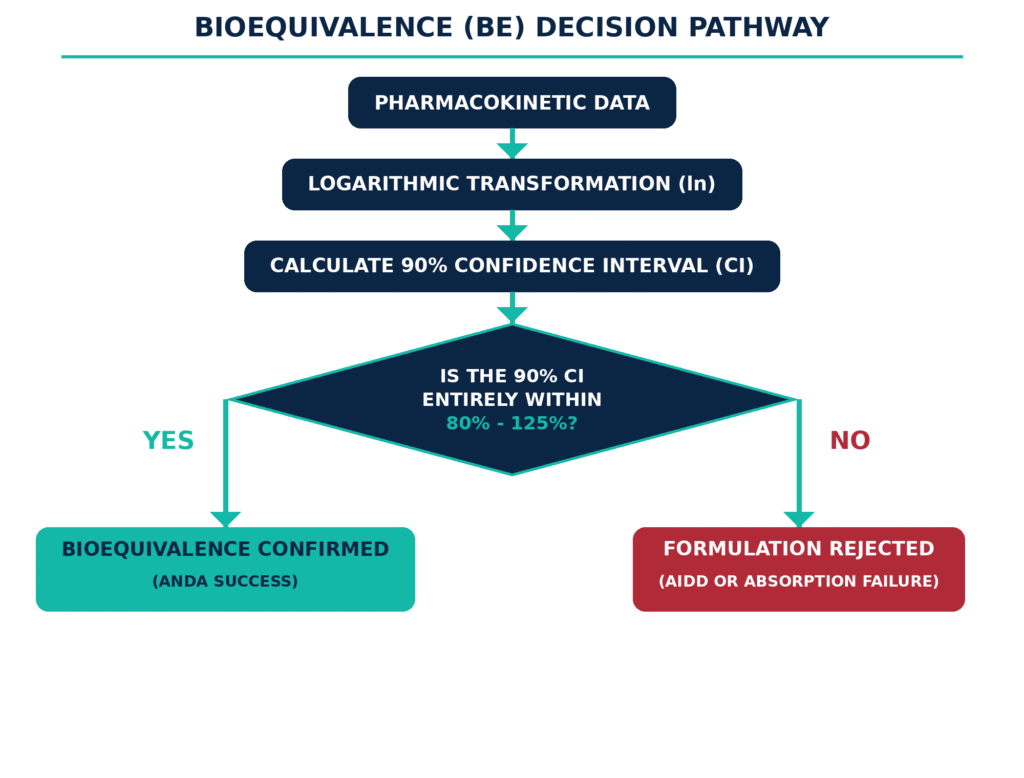
Bioequivalence for modified-release formulations should be established through carefully designed in vivo clinical studies. Crossover trials, especially the two-period, two-sequence, two-treatment, single-dose crossover design, are widely used because each participant receives both the test and reference products and therefore serves as their own control. This approach helps minimize the impact of between-subject pharmacokinetic variability. For highly variable drugs, defined as products with an intra-subject coefficient of variation greater than 30% for Cmax or AUC, a fully replicated crossover design with three- or four-period treatment sequences may be appropriate. Such a design enables bioequivalence limits to be scaled based on the variability observed with the Reference Listed Drug.
Critical pharmacokinetic sampling parameters for modified-release bioequivalence studies include:
Fasting vs. Fed Studies: Bioequivalence programs must assess the effect of food because food may delay gastric emptying or interact physically with the polymer matrix, thereby changing the drug-release behavior. Fasting studies generally require an 8-hour fast before dosing. Fed studies use a standardized high-fat, high-calorie meal containing approximately 800 to 1000 kcal, with approximately 50% to 60% of total calories derived from fat, consumed 30 minutes before administration of the drug product.
Sampling Schedule: Pharmacokinetic sampling should generally continue for a duration covering at least 5 to 7 elimination half-lives to capture more than 80% to 90% of the total AUC. Frequent sampling during the early post-dose period, particularly within approximately 5 to 15 minutes after administration, is important for avoiding “first-point Cmax” errors. These errors can occur when the first measured plasma concentration is also the highest observed concentration, indicating that the early absorption phase may not have been adequately characterized.
Analyte Selection: The parent drug should be quantified in systemic circulation because its concentration-time profile is highly responsive to the release characteristics of the formulation. Metabolites are generally included in the analysis only when they are produced through pre-systemic (first-pass) metabolism and make a meaningful contribution to the product’s safety or efficacy profile.
Endogenous Compounds: For drugs that are naturally present in the human body, including certain hormones or electrolytes, baseline plasma concentrations must be determined at multiple time points before dosing. These baseline values are then subtracted from post-administration concentrations to distinguish the absorption attributable to the administered formulation.
Enantiomers: Chiral assays may be necessary when the active enantiomer demonstrates highly non-linear pharmacokinetics or possesses a substantially different safety-efficacy profile compared with the inactive enantiomer.
For insights into the rigorous characterization techniques required for modern metabolic therapies, read our deep dive on GLP-1 peptide analytical characterization.
Analytical and Formulation Milestones in CDMO Services for Generic Oral Solid Dosage Forms
Establishing therapeutic equivalence for generic solid oral products depends on the coordinated execution of phase-appropriate analytical validation and reliable formulation scale-up. This integrated approach helps ensure that the physical and chemical properties of the generic product remain comparable to those of the RLD throughout the intended shelf-life.
To achieve readiness for ANDA submission, developers must complete a sequence of closely controlled technical and regulatory milestones. During pre-formulation, the API’s solid-state chemistry is characterized through the evaluation of polymorphism, hygroscopicity, particle size distribution, and compatibility with the intended excipients. As development progresses from laboratory-scale prototypes to pilot-scale batches, analytical methods must be validated in accordance with International Council for Harmonisation (ICH) guidelines. This process includes the validation of stability-indicating high-performance liquid chromatography (HPLC) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods capable of detecting and quantifying degradation products, process impurities, and genotoxic or nitrosamine compounds at trace-level concentrations.
ResolveMass Laboratories Inc. supports these essential development stages through the application of advanced mass spectrometry technologies, including HRMS and LC-MS/MS, together with automated dissolution profiling to develop highly discriminating quality control methods. These advanced analytical systems help verify that each batch demonstrates accurate content uniformity, strong physical stability, and a dissolution profile that remains superimposable with that of the RLD.
| Development Phase | Core Analytical Milestones | Core Formulation Milestones | Primary Regulatory Documentation Generated |
|---|---|---|---|
| Pre-Formulation & Compatibility | Polymorph characterization, pKa and solubility profiling, API-excipient compatibility assays. | Excipient screening, selection of “spring and parachute” polymers for poorly soluble APIs. | Compatibility study reports, Solid-State characterization files, initial target product profiles. |
| Prototype & Process Optimization | Dissolution method development, initial impurity profiling, baseline stability runs. | Granulation trial execution, compaction force mapping, encapsulation trial runs. | Discrimination power reports for dissolution, initial prototype batch records. |
| Pilot Scale-Up & BE Batch Manufacture | Validation of analytical and bioanalytical methods, accelerated and long-term stability initiation. | Execution of GMP pilot batches (minimum 1/10th commercial scale or 100,000 units). | Executed GMP batch records, complete analytical validation reports, 6-month stability datasets. |
| ANDA Filing & Submission | Compiling full analytical validation packages, final impurity profiling, forced degradation reports. | Critical process parameter (CPP) definition, commercial scale-up process validation. | Complete eCTD Module 3 (CMC section), Drug Master File (DMF) Letters of Authorization. |
Review a real-world application of these regulatory validation milestones in our peptide characterization case study of semaglutide.
Understanding the Regulatory Shift in Bioequivalence: The ICH M13A Impact
The harmonization introduced through the final ICH M13A guideline issued in October 2024 simplifies the global registration of immediate-release solid oral dosage forms by standardizing bioequivalence study requirements across multiple regulatory regions. One of the most important changes is the elimination of the routine requirement for separate fasting and fed studies for non-high-risk products. Instead, a single highly sensitive study, typically conducted under fasting conditions, may be sufficient.
Before the implementation of the ICH M13A guidelines, regulatory agencies, including the FDA, Health Canada, and the EMA, differed considerably in their expectations for food-effect evaluation. Generic drug developers were frequently required to conduct separate fasting and fed in vivo clinical trials to obtain marketing authorization in different jurisdictions. This duplication increased both development timelines and overall costs.
For companies navigating these updated frameworks in the North American market, partnering with an established CDMO for generic drug development in Canada ensures comprehensive regulatory and logistical alignment.
Under the standardized, risk-based approach established by the ICH M13A framework:
Non-High-Risk Products: A single in vivo bioequivalence study conducted under the most sensitive physiological condition, typically the fasting state, is sufficient to demonstrate therapeutic equivalence, regardless of the food-related instructions included in the RLD’s clinical labeling.
High-Risk Products: Products developed using specialized technologies to intentionally optimize a food effect or improve solubility, such as specific micro-emulsions or high-energy amorphous dispersions, are categorized as high-risk products. These products continue to require both fasting and fed in vivo studies.
Industry Economic Impact: In October 2024, the FDA revised more than 800 oral immediate-release Product-Specific Guidances (PSGs) to align with M13A. This change was estimated to generate annual industry savings of 50 million dollars by removing the need for certain unnecessary clinical trials.
The ICH M13 Roadmap includes three broader tiers:
ICH M13A: Establishes study designs and data analysis principles for immediate-release formulations.
ICH M13B: Drafted in February 2025, this guideline describes scientific principles and biowaiver considerations for additional strengths within a product line.
ICH M13C: Addresses highly variable drugs, Narrow Therapeutic Index (NTI) drugs, and highly complex clinical study designs.
Efficiently executing these technical studies requires experienced oversight; see how our targeted support channels can advance your CDMO for generic projects in Canada.
Post-Approval Changes and SUPAC-MR Regulatory Pathways
Post-approval changes involving the composition, manufacturing site, or manufacturing process of modified-release products are governed by the FDA’s SUPAC-MR guidelines. These requirements are intended to ensure that approved changes do not negatively affect product quality or drug-release performance. Such modifications are categorized as Level 1 (minor), Level 2 (moderate), or Level 3 (major), with each category requiring a defined level of dissolution testing and, where applicable, clinical bioequivalence data.
Under SUPAC-MR, the significance of an excipient change is determined primarily by whether the excipient performs a functional role in controlling drug release or serves a non-functional role. For non-release-controlling excipients, including binders, fillers, and disintegrants, changes are generally managed as follows:
Level 1 (Minor): Composition changes that remain within narrowly defined limits, such as filler changes of 5% w/w or less of the total core weight, generally require only annual report filing. One batch must also be placed on long-term stability.
Level 2 (Moderate): Changes in excipient quantities that exceed Level 1 limits but remain up to a threshold of 10% w/w, as well as changes in the technical grade or specification of an excipient, such as changing from coarse to granular binders, require submission of a Changes Being Effected (CBE) supplement or Prior Approval Supplement (PAS). Supporting evidence must include long-term stability data and multipoint comparative dissolution profiles collected at 15, 30, 45, 60, and 120 minutes in three distinct media, such as pH 1.2, 4.5, and 6.8.
Level 3 (Major): Qualitative or quantitative changes to excipients that exceed 10% w/w require a full Prior Approval Supplement (PAS). The submission must be supported by comprehensive chemistry, manufacturing, and controls (CMC) documentation, accelerated stability data, and either a complete in vivo clinical bioequivalence study or a validated in vitro-in vivo correlation (IVIVC) surrogate.
For release-controlling excipients, such as functional ethylcellulose or HPMC coatings, any change of 10% w/w or less in the total polymer content is considered a Level 2 change. Such a change requires a PAS supported by multipoint dissolution profiles generated in multiple physiological media. Any modification exceeding 10% w/w of the functional polymer content is classified as a Level 3 change and automatically requires a clinical biostudy or a validated IVIVC.
Managing formulation modifications while preserving compliance requires a specialized generic pharmaceutical CDMO in Canada equipped for lifecycle management.
Mathematical Surrogation: Establishing In Vitro-In Vivo Correlation (IVIVC)
An In Vitro-In Vivo Correlation (IVIVC) is a predictive mathematical model that establishes a direct and quantitative relationship between the laboratory dissolution behavior of a drug product and its clinical pharmacokinetic response. Once appropriately validated, a Level A IVIVC can serve as a regulatory surrogate for human bioequivalence studies and may allow developers to obtain biowaivers for certain post-approval manufacturing changes.
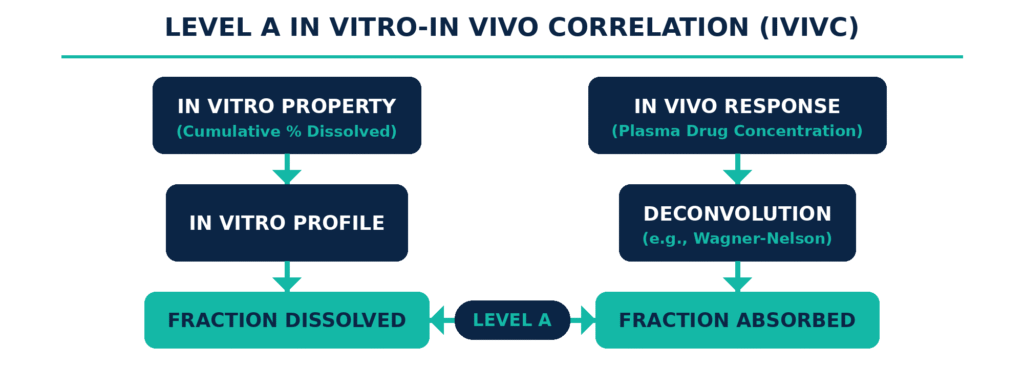
Developing an IVIVC is particularly valuable for modified-release formulations because it provides a quantitative method for assessing how changes in formulation composition, manufacturing processes, or manufacturing sites may affect in vivo drug absorption. To establish an effective correlation, formulation scientists apply mathematical approaches across three distinct stages. First, a functional relationship is developed between the in vitro input and the in vivo output. Next, a structural model is established using clinical data generated from multiple formulations with different release rates, typically demonstrating at least a 10% difference in dissolution. Finally, the model is parameterized to enable prediction of plasma concentration profiles.
The regulatory applicability and complexity of IVIVC models are described through four correlation levels:
Level A: This represents a direct, point-to-point relationship between in vitro dissolution and the in vivo input rate, which reflects the fraction of drug absorbed over time. Establishing this model requires a deconvolution procedure, such as the Wagner-Nelson method or numerical deconvolution, to derive the absorption profile from the in vivo plasma concentration-time curve. The resulting absorption profile is then directly compared with the fraction of drug dissolved. Level A is the only IVIVC category directly accepted by the FDA for supporting biowaivers for major (Level 3) post-approval changes or for waiving clinical studies for additional strengths of an approved drug line.
Level B: This correlation method applies statistical moment analysis to compare the mean in vitro dissolution time (MDT in vitro) with the corresponding mean in vivo residence time or dissolution time. Since the analysis is based on summarized statistical parameters rather than a complete point-by-point comparison of the profiles, distinct plasma concentration–time curves may still produce identical mean residence values. As a result, Level B correlations generally have limited value for supporting regulatory submissions.
Level C: This type of correlation establishes a relationship between a single in vitro dissolution parameter, such as the time required for 50% of the drug to dissolve (t50%), and one pharmacokinetic parameter, such as Cmax or AUC. Because it evaluates only one dissolution metric against one pharmacokinetic outcome, it does not capture the complete shape of the plasma concentration–time profile. Therefore, Level C correlations are generally insufficient to support regulatory biowaivers.
Multiple Level C: This approach correlates one or more relevant PK parameters with the cumulative fraction dissolved at multiple discrete time points representing the early, middle, and late stages of dissolution. It is considered to have utility comparable to a Level A correlation. However, when a Multiple Level C correlation can be established, formulation scientists can generally progress toward developing a complete Level A model.
Strategically mitigating development risks is central to successful commercialization when outsourcing generic drug development in Canada.
Quality by Design (QbD) Framework and Advanced Analytical Infrastructure
Developing generic solid oral formulations within a Quality by Design (QbD) framework ensures that product and process quality are scientifically and systematically incorporated from the beginning rather than assessed only after manufacturing has been completed. This approach establishes relationships between Critical Material Attributes (CMAs), Critical Process Parameters (CPPs), and the product’s Critical Quality Attributes (CQAs) to define a scientifically justified and compliant Design Space.
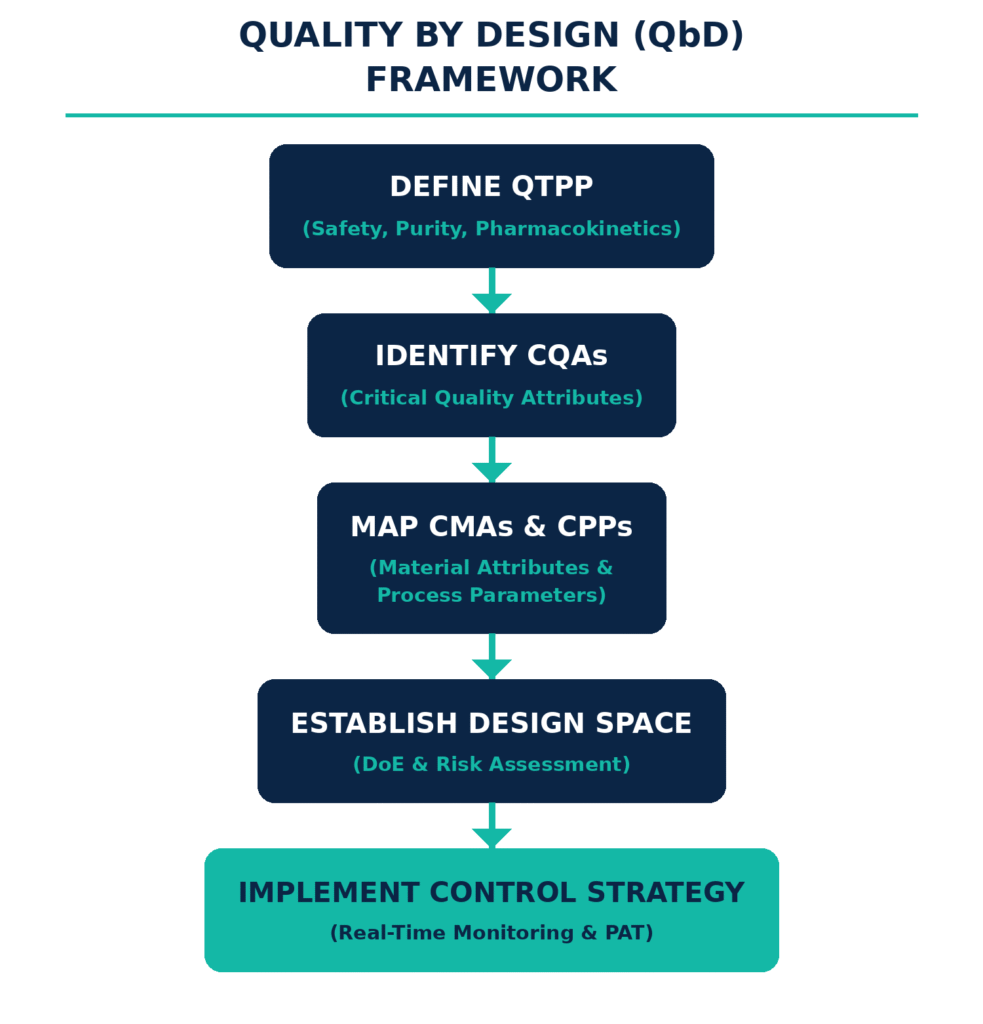
The QbD methodology begins by defining the Quality Target Product Profile (QTPP), which establishes the desired product characteristics, including dosage strength, disintegration kinetics, dissolution velocity, and in vivo bioavailability. Through structured risk assessment techniques, such as Failure Mode and Effects Analysis, developers identify the critical material attributes, including API particle size distribution, crystal habit, and excipient polymer viscosity, as well as critical process parameters, such as granulator impeller speed, fluid-bed drying temperature, and tablet compression force, that may influence the final product CQAs.
Using Design of Experiments (DoE), formulation scientists conduct multivariate studies to characterize the multidimensional relationships among these variables. This process enables the definition of the “Design Space,” within which the manufacturer can operate with a high degree of flexibility because process variation occurring within the established boundaries has been demonstrated to produce a product that meets predefined quality and therapeutic requirements.
To confirm the suitability of this Design Space and prepare a successful ANDA dossier, developers must utilize advanced analytical infrastructure. ResolveMass Laboratories Inc. supports these technical requirements through specialized analytical services, including high-performance liquid chromatography (HPLC) for accurate assay determination, liquid chromatography-tandem mass spectrometry (LC-MS/MS) for ultra-trace impurity profiling, including nitrosamines and genotoxic compounds, and high-resolution mass spectrometry (HRMS) for comprehensive polymorph and degradation studies. In addition, discriminating dissolution systems are used to evaluate formulations under a range of physiological conditions. These capabilities help ensure that generic tablets or capsules release their active ingredients with maximum reproducibility while remaining aligned with global compendial standards and regulatory expectations.
To secure submission-ready validation datasets that satisfy these complex multi-variant parameters, leverage premium analytical development for generic drugs in Canada.
Conclusion
Partnering with a specialized contract organization for CDMO Services for Generic Oral Solid Dosage Forms represents a highly secure strategy for pursuing ANDA approval and achieving long-term commercial viability. This collaborative approach brings together advanced polymer chemistry, state-of-the-art mass spectrometry, and detailed knowledge of global regulatory requirements to address the complexities associated with tablets, capsules, and modified-release drug delivery systems. Whether the challenge involves managing the physical limitations of cohesive powder flow during high-speed capsule encapsulation, designing matrix tablets capable of resisting alcohol-induced dose dumping, or validating point-to-point Level A IVIVC models, generic drug development programs must be based on rigorous, scientifically supported, and data-driven principles.
Regulatory developments such as the risk-based ICH M13A guideline are helping streamline global registration pathways while also requiring analytical and formulation strategies to be optimized from the earliest stages of pre-formulation through scale-up. Aligning development activities with a systematic Quality by Design (QbD) framework ensures that process variables remain controlled within a validated Design Space, thereby reducing the risk of manufacturing failures and Complete Response Letters (CRLs). ResolveMass Laboratories Inc. serves as an authoritative technical partner in this environment, offering advanced chromatography, mass spectrometry, and validation expertise required to develop regulatory-grade CMC dossiers and support the delivery of safe, effective, and compliant generic therapies to global markets.
For advanced analytical support and regulatory-grade dossiers, reach out to the technical team at ResolveMass Laboratories Inc. via our Contact us page.
Frequently Asked Questions (FAQs)
Under the ICH M9 guideline, a BCS-based biowaiver may be considered for certain highly soluble immediate-release solid oral dosage forms belonging to BCS Class I or BCS Class III. The eligibility criteria require the product to meet the applicable requirements for solubility, permeability, dissolution, and formulation composition. Modified-release products, Narrow Therapeutic Index (NTI) drugs, and dosage forms intended for absorption within the oral cavity are not eligible for this type of biowaiver.
Under SUPAC-MR, the FDA categorizes post-approval modifications into three levels based on their potential impact on product performance. Level 1 changes are minor and generally have limited potential to affect product quality, while Level 2 changes involve moderate modifications and may require CBE or PAS documentation with comparative dissolution data. Level 3 changes are major modifications, such as substantial changes to release-controlling polymer content, and may require a PAS supported by in vivo bioequivalence data or a validated IVIVC.
Dose dumping occurs when a modified-release formulation releases a large portion or the entire drug dose much faster than intended, potentially producing excessive systemic exposure and toxicity. In generic modified-release development, this risk is commonly assessed through comparative in vitro dissolution testing under alcohol-containing conditions. Testing may involve 12 dosage units in 0.1 N HCl (pH 1.2) containing increasing ethanol concentrations of 0%, 5%, 20%, and 40% v/v over a 2-hour period.
High-potency active pharmaceutical ingredients (HPAPIs), including cytotoxics and certain hormones, require specialized development and manufacturing strategies because even very small quantities may pose significant occupational or cross-contamination risks. Their handling may require high-containment technologies, dedicated or isolated manufacturing areas, and carefully controlled environmental conditions. These controls are essential for protecting personnel, preventing cross-contamination, and meeting stringent cGMP requirements for high-containment processing.
The final ICH M13A guideline, implemented in October 2024, introduced a more harmonized and risk-based approach to bioequivalence requirements for immediate-release solid oral dosage forms. For non-high-risk products, developers may no longer routinely need separate fasting and fed in vivo clinical studies. Instead, a single study conducted under the most sensitive physiological condition, typically fasting conditions, may be sufficient to establish bioequivalence.
A Drug Master File (DMF) is a confidential regulatory submission containing detailed chemistry, manufacturing, and controls (CMC) information related to materials such as an active pharmaceutical ingredient, excipient, or packaging component. A CDMO may prepare and maintain the DMF and provide the client with a Letter of Authorization (LOA). This authorization allows regulatory authorities to review the confidential DMF information as part of the client’s ANDA without requiring the CDMO to disclose proprietary information directly to the applicant.
High-speed capsule filling can be affected by poor powder flow, strong interparticle cohesion, electrostatic charge accumulation, and segregation within the hopper or feeding system. These issues can cause powder bridging, inconsistent die filling, and variation in capsule fill weight. CDMOs address these challenges through appropriate excipient selection, optimization of glidant levels, wet or dry granulation when necessary to improve bulk density and flow, and careful control of relative humidity within the manufacturing environment.
Reference:
- Wang, R. (2022, August 18). Bioequivalence studies for generic drug development [PowerPoint slides]. U.S. Food and Drug Administration. FDA source
- U.S. Food and Drug Administration. (2014, May). ANDAs: Stability testing of drug substances and products: Questions and answers: Guidance for industry. U.S. Department of Health and Human Services. FDA guidance document
- European Medicines Agency. (2025, January 25). ICH Guideline M13A on bioequivalence for immediate-release solid oral dosage forms—Scientific guideline. EMA guideline page
- Zhang, L. (2024, September 25). ICH M13A: First ICH guideline for bioequivalence [PowerPoint slides]. U.S. Food and Drug Administration. FDA presentation
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2025, February 24). M13B: Bioequivalence for immediate-release solid oral dosage forms: Additional strengths biowaiver (Draft version). U.S. Food and Drug Administration. FDA guidance document
- Pund, S., Dhande, M., Jayatpal, S., Tupe, A., et al. (2024). Scale-up and postapproval changes (SUPAC) guidelines for industry: A comprehensive review. Multidisciplinary Reviews, 7(4), 2024071. https://doi.org/10.31893/multirev.2024071

