
Introduction
Selecting the right partner is a critical step in generic drug development. Pharmaceutical companies must Choose Right CRO/CDMO for ANDA Submission to ensure that bioequivalence studies, analytical testing, regulatory documentation, and manufacturing data meet FDA requirements for Abbreviated New Drug Applications (ANDA). When development activities are aligned with regulatory expectations from the beginning, the chances of smooth approval increase.
An experienced CRO/CDMO can guide companies through complex development and regulatory processes. Their scientific expertise, regulatory knowledge, and infrastructure help manage formulation development, analytical testing, and clinical studies efficiently. In many cases, they work as an extension of the sponsor’s internal research and regulatory teams. Choosing an unsuitable partner can lead to failed studies, regulatory deficiency letters, and delays in product approval. These issues can increase development costs and reduce market opportunities in competitive generic drug markets. Therefore, companies must carefully evaluate partners before making a decision.
Explore our specialized support for your submission: Analytical Requirements for ANDA Submissions
This article explains how companies can Choose Right CRO/CDMO for ANDA Submission by evaluating key factors such as regulatory expertise, analytical capabilities, quality systems, and manufacturing readiness. Understanding these criteria helps pharmaceutical organizations build reliable partnerships and improve the chances of successful ANDA approval.
Share via:
Quick Summary (Key Takeaways)
- Selecting the right CRO/CDMO for ANDA submission directly impacts approval timelines, regulatory compliance, and development costs.
- Evaluate ANDA experience, regulatory track record, and FDA inspection history before partnering.
- Ensure the CRO/CDMO has bioequivalence, analytical, formulation, and regulatory capabilities aligned with your product.
- Assess technology transfer, scale-up manufacturing, and documentation readiness for FDA audits.
- Strong project management, transparency, and communication systems reduce delays during ANDA review cycles.
- Verify quality systems (cGMP, GLP, GCP) and data integrity practices to prevent regulatory setbacks.
- Review facility readiness, equipment capability, and capacity for commercial scale manufacturing.
- A strategic CRO/CDMO partner should help optimize ANDA dossier preparation, reduce deficiency letters, and accelerate approval.
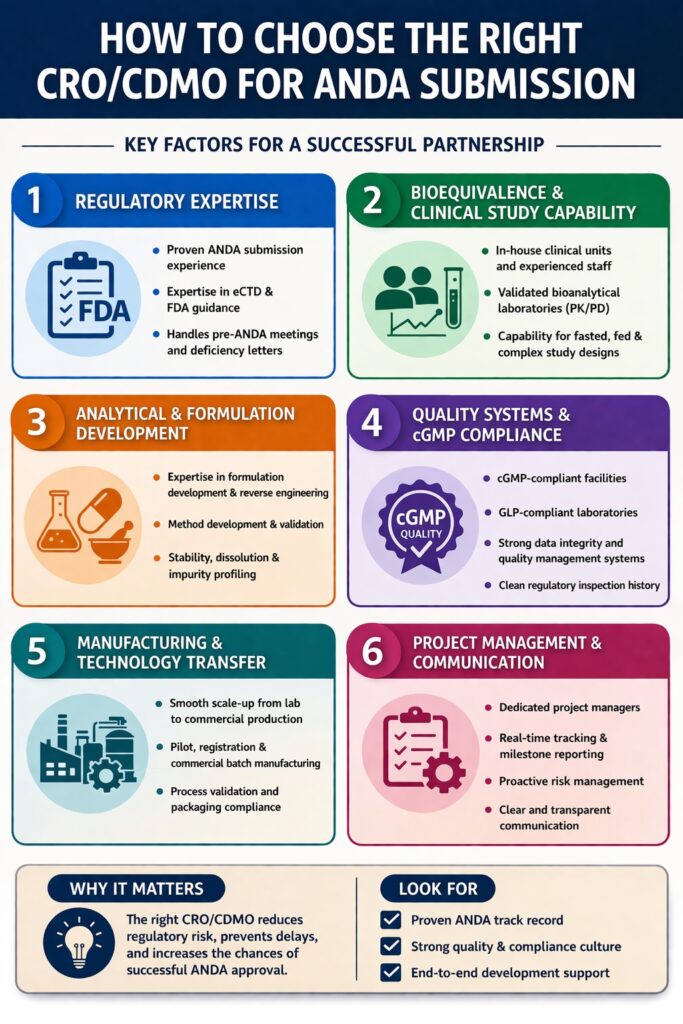
Key Criteria to Choose Right CRO/CDMO for ANDA Submission
When companies plan to Choose Right CRO/CDMO for ANDA Submission, they should focus on a combination of regulatory knowledge, analytical capabilities, manufacturing readiness, and strong quality systems. These factors ensure that development activities remain aligned with regulatory expectations and scientific standards.
A CRO/CDMO should also be capable of integrating product development with regulatory strategy. This means formulation design, analytical method development, and manufacturing planning should all support the final ANDA submission requirements. Early alignment between development and regulatory strategy helps prevent costly delays or additional data requests later.
Learn more about the benefits of expert partnership: Outsourcing Analytical Testing for ANDA Submissions
Below is a structured evaluation framework used by many pharmaceutical organizations when they Choose Right CRO/CDMO for ANDA Submission.
| Evaluation Factor | Why It Matters for ANDA Submission |
|---|---|
| ANDA submission experience | Reduces regulatory risk and improves dossier quality |
| Bioequivalence study capability | Essential for demonstrating therapeutic equivalence |
| Regulatory expertise | Helps prepare FDA-compliant documentation |
| Analytical and formulation capabilities | Ensures reproducibility and product stability |
| cGMP compliance and quality systems | Prevents regulatory observations |
| Technology transfer capability | Supports scale-up and commercialization |
| Inspection history | Indicates regulatory reliability |
Using a structured framework helps companies compare CRO/CDMO partners objectively. It allows decision-makers to assess scientific expertise, regulatory reliability, and operational capabilities before finalizing a development partnership.
Regulatory Expertise is the First Factor When You Choose Right CRO/CDMO for ANDA Submission
Regulatory expertise is often the first indicator when companies Choose Right CRO/CDMO for ANDA Submission. A CRO/CDMO with strong regulatory knowledge understands FDA expectations for generic drug development and ANDA documentation.
Experienced regulatory teams help ensure that submission components are organized correctly and comply with current regulatory guidelines. Their understanding of regulatory procedures also allows them to guide sponsors through complex submission requirements and communication with regulatory authorities.
Ensure your methods meet the gold standard: ANDA Method Validation Requirements
A CRO/CDMO should demonstrate experience in:
- Preparing Module 2–5 sections of the eCTD format
- Managing FDA pre-ANDA meetings
- Handling deficiency letters and Complete Response Letters (CRLs)
- Supporting bioequivalence protocol approvals
- Preparing drug master file (DMF) cross-references
These capabilities ensure that regulatory documentation is properly structured and scientifically justified. Clear and organized submissions often lead to more efficient regulatory review cycles.
Organizations that frequently interact with regulatory agencies are also better prepared to anticipate regulatory expectations. Their experience allows them to identify potential issues early and resolve them before submission.
Key indicators to review include:
- Number of successful ANDA approvals supported
- Experience with the 505(j) regulatory pathway
- Familiarity with USFDA guidance for generic drugs
Research suggests that regulatory outsourcing to specialized CROs improves coordination and compliance with FDA documentation standards (Xiaofang et al., 2018). Dedicated regulatory teams also monitor evolving regulatory guidance to ensure submissions remain compliant.
Bioequivalence and Clinical Study Capability
Bioequivalence studies are a central requirement for most generic drug approvals. These studies demonstrate that the generic drug performs the same way as the reference listed drug (RLD) in the human body.
When companies Choose Right CRO/CDMO for ANDA Submission, they must confirm that the partner has strong bioequivalence and clinical study capabilities. These studies require specialized clinical infrastructure, experienced investigators, and validated analytical methods.
Access comprehensive support for your clinical data: Bioanalytical Services
A suitable CRO/CDMO should provide:
- Clinical pharmacology units
- PK/PD analysis expertise
- Validated bioanalytical laboratories
- Healthy volunteer study management
Additional important considerations include:
- Ability to conduct fasted and fed bioequivalence studies
- Experience with highly variable drugs
- Capability to conduct replicate crossover study designs
CROs with advanced bioanalytical laboratories reduce the risk of study failure or data rejection. Their validated analytical methods ensure accurate pharmacokinetic analysis and reliable regulatory data.
Experienced clinical teams also ensure that studies follow ethical guidelines and regulatory standards. This combination of clinical infrastructure and analytical expertise significantly improves bioequivalence study success rates.
Analytical and Formulation Development Capabilities
Strong analytical and formulation capabilities are essential when companies Choose Right CRO/CDMO for ANDA Submission. These capabilities help create a generic formulation that matches the reference listed drug in quality, performance, and stability.
Generic drugs must match the RLD in several aspects:
- Dosage form
- Strength
- Route of administration
- Bioavailability
- Quality attributes
A competent CRO/CDMO should provide comprehensive formulation development services such as:
Formulation development services
- Reverse engineering of RLD
- Excipient compatibility studies
- Dissolution profile matching
- Stability optimization
Analytical capabilities
- Method development and validation
- Impurity profiling
- Stability testing
- Extractables and leachables analysis
Identify and quantify impurities with precision: Impurity Identification for ANDA Submission
These services ensure that the final product meets FDA quality expectations and pharmacopeial standards. Advanced analytical tools also allow scientists to detect potential formulation challenges early.
By identifying formulation issues early in development, companies can adjust product design before regulatory submission. This proactive approach improves product consistency and regulatory acceptance.
Mitigate risks with dedicated contaminant analysis: Nitrosamine Testing in ANDA Submissions
Quality Systems and cGMP Compliance
Quality systems play a major role when organizations Choose Right CRO/CDMO for ANDA Submission. Strong quality management frameworks ensure that development, testing, and manufacturing activities follow regulatory standards.
Regulatory authorities closely evaluate data integrity, documentation accuracy, and quality systems during inspections. CRO/CDMOs must therefore implement strict quality assurance procedures and compliance controls.
A strong quality framework ensures:
- Accurate data generation
- Regulatory compliance
- Successful inspections
Key quality indicators include:
- cGMP-compliant manufacturing facilities
- GLP-compliant analytical laboratories
- Data integrity controls
- Electronic documentation systems
Address potential gaps before they become issues: ANDA Analytical Deficiencies
Sponsors should also review inspection history from agencies such as:
- US FDA
- EMA
- Health Canada
- MHRA
A strong inspection history indicates reliable regulatory compliance and well-maintained quality systems. CRO/CDMOs with consistent inspection success usually maintain disciplined operational practices.
Research on pharmaceutical outsourcing also highlights that quality systems and compliance frameworks are major determinants of regulatory approval success (Lakings & Lakings, 2013).
Technology Transfer and Manufacturing Scale-Up
ANDA approval requires more than development work. The product must also be ready for commercial manufacturing after approval.
When companies Choose Right CRO/CDMO for ANDA Submission, they should ensure the partner has strong technology transfer and manufacturing scale-up capabilities. These capabilities allow processes developed in the laboratory to be reproduced at commercial production scale.
Critical capabilities include:
- Pilot scale manufacturing
- Process validation
- Commercial batch manufacturing
- Packaging and labeling compliance
| Development Stage | Required Capability |
|---|---|
| Formulation development | Lab-scale batch development |
| Scale-up | Pilot scale manufacturing |
| Registration batch | cGMP manufacturing |
| Commercial supply | Large scale production |
Organizations that offer integrated CRO and CDMO services often streamline the transition from development to manufacturing. Integrated service models reduce coordination challenges between multiple vendors.
This integration improves process consistency and helps accelerate product launch timelines.
Project Management and Communication Systems
Even highly skilled CRO/CDMO organizations may struggle if project management is weak. Effective project management ensures that development tasks, documentation preparation, and regulatory submissions stay on schedule.
When companies Choose Right CRO/CDMO for ANDA Submission, they should evaluate the partner’s project management systems and communication processes.
Important features include:
- Dedicated project managers
- Real-time project tracking
- Risk management systems
- Transparent milestone reporting
Strong project management helps prevent:
- Protocol deviations
- Documentation delays
- Regulatory submission errors
Research on generic drug development indicates that strong coordination between sponsors and CRO teams significantly improves project timelines and regulatory outcomes (Xiaofang et al., 2018).
Facility Infrastructure and Technology Capabilities
Facility infrastructure is another important factor when companies Choose Right CRO/CDMO for ANDA Submission. Advanced infrastructure allows CRO/CDMOs to handle complex formulations and specialized drug delivery systems.
Modern pharmaceutical development requires sophisticated analytical instruments and controlled manufacturing environments. CRO/CDMOs with modern facilities can support both routine and complex development programs.
Important capabilities include:
- High-potency drug handling
- Controlled environment manufacturing
- Advanced analytical instruments
- Stability chambers meeting ICH guidelines
Sponsors should also evaluate whether facilities support:
- Modified release formulations
- Complex injectables
- Combination products
Conduct thorough safety evaluations for generic products: Nitrosamine Risk Assessment in Generic Drugs
Facilities built to meet modern regulatory expectations help ensure smoother inspections and regulatory approvals.
Cost Transparency and Long-Term Value
Cost is an important factor, but selecting the cheapest partner is not always the best decision. Companies should focus on long-term value when they Choose Right CRO/CDMO for ANDA Submission.
Lower-cost providers may lack the infrastructure or regulatory expertise required for complex ANDA projects. This may lead to study failures, redevelopment work, or regulatory delays.
Instead, companies should evaluate:
- Cost versus expertise
- Regulatory track record
- Development timelines
- Quality assurance systems
A capable CRO/CDMO can help reduce:
- Bioequivalence study failures
- Repeated formulation development
- Regulatory deficiency cycles
Generic drug development can cost between $1 million and $5 million depending on complexity (Parasrampuria et al., 2021). Investing in an experienced partner often leads to faster approvals and better return on investment.
Red Flags to Avoid When Choosing a CRO/CDMO
Companies should avoid CRO/CDMO partners that show the following warning signs:
- Limited ANDA experience
- Poor FDA inspection history
- Lack of validated analytical methods
- Weak data integrity systems
- Poor communication and project transparency
These warning signs often indicate operational or regulatory weaknesses that may affect development quality. Ignoring these issues can result in regulatory deficiencies and project delays.
Thorough due diligence is therefore important when selecting a development partner. Sponsors should review regulatory records, conduct facility audits, and examine previous project outcomes.
Taking these steps helps companies identify reliable partners and avoid costly development risks.
Conclusion
Choosing the right development partner is essential for successful generic drug approval. Pharmaceutical companies must carefully Choose Right CRO/CDMO for ANDA Submission to ensure regulatory compliance, efficient development, and smooth commercialization.
The ideal CRO/CDMO should demonstrate strong regulatory expertise, proven bioequivalence study capability, advanced analytical infrastructure, robust quality systems, and scalable manufacturing facilities. These capabilities ensure that development activities meet both scientific and regulatory expectations.
Companies that evaluate partners based on ANDA experience, regulatory compliance history, and integrated development capabilities significantly increase their chances of successful FDA approval.
A strong CRO/CDMO partnership supports not only ANDA dossier preparation but also long-term product commercialization. Effective collaboration between sponsors and development partners leads to faster development timelines and improved regulatory outcomes.
For consultation or project discussion, visit: Contact page
Reference:
- U.S. Food and Drug Administration. (2022). Good ANDA submission practices: Guidance for industry. https://www.fda.gov/media/187318/download
- Liu, Q., Davit, B. M., Cherstniakova, S. A., Dandamudi, S., Walters, J. F., Lee, C. H., Raines, K. W., Ren, K., Williamson, L. N., & Conner, D. P. (2011). Common deficiencies with bioequivalence submissions in abbreviated new drug applications assessed by FDA. AAPS PharmSciTech, 14(1), 19–22. https://doi.org/10.1208/s12248-011-9312-7
- Pawar, J., Hegde, N., & Sharma, S. (2025). Focusing on first cycle approval in ANDA submission: Understanding common deficiencies & case study insights. Therapeutic Innovation & Regulatory Science, 59(3), 426–437. https://doi.org/10.1007/s43441-025-00755-5

