
🔹 Executive Summary
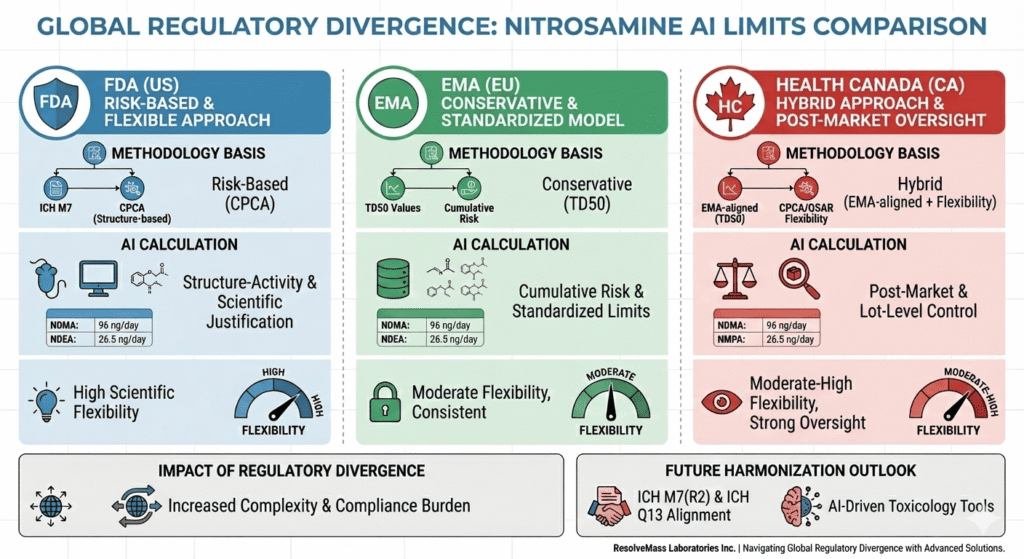
- The Nitrosamine AI limits comparison among FDA, EMA, and Health Canada reveals key regulatory divergences in analytical approaches, acceptable intake calculations, and lifecycle control expectations.
- FDA applies a structure-based approach rooted in ICH M7 and CPCA methodology, emphasizing risk communication flexibility.
- EMA prioritizes consistency and cumulative risk, incorporating Total Daily Intake and structural read-across principles.
- Health Canada aligns closely with EMA but mandates post-market surveillance and lot-level AI compliance verification.
- Divergence impacts global drug registration, bioequivalence bridging, and quality documentation harmonization.
- Future regulatory harmonization through ICH Q13 and AI-based predictive toxicology could unify risk assessment paradigms.
Introduction: Understanding the Nitrosamine AI Limits Comparison
When conducting a Nitrosamine AI limits Comparison across major regulatory authorities, clear differences in regulatory philosophy become apparent. While the FDA, EMA, and Health Canada all share the responsibility of safeguarding public health, they apply different methods to assess, quantify, and manage nitrosamine-related risks.
👉 Learn more about comprehensive nitrosamine analysis approaches
These differences influence acceptable intake thresholds, interpretation of animal carcinogenicity data, and how uncertainty is handled. As a result, pharmaceutical manufacturers operating in multiple regions face added regulatory complexity. A single nitrosamine impurity may trigger different compliance actions in different markets, increasing the burden on dossier preparation, bioequivalence justification, and long-term lifecycle management.
👉 Explore a structured risk assessment framework.
Looking for Health Canada’s official acceptable intake limits for nitrosamine impurities?
Access Health Canada’s published AI limits and regulatory expectations to support compliant risk assessments and post-market oversight.
👉 Review Health Canada’s established nitrosamine AI limits
Need EMA-approved acceptable intake values for specific N-nitrosamines?
Download the EMA’s official Appendix 1 spreadsheet detailing compound-specific AI limits used across EU regulatory submissions.
👉 Access EMA’s acceptable intake list for N-nitrosamines
Want clarity on FDA’s acceptable intake limits for nitrosamine impurities?
Explore FDA CDER’s guidance outlining AI limits, risk assessment principles, and structure-based evaluation approaches.
👉 View FDA guidance on nitrosamine acceptable intake limits
1. FDA’s Framework for Nitrosamine AI Limits Comparison
The FDA determines nitrosamine AI limits using a risk-based framework under ICH M7, primarily through the Carcinogenic Potency Categorization Approach (CPCA).
The FDA’s model emphasizes scientific understanding and structural similarity, especially when direct carcinogenicity data are limited. Instead of relying only on fixed default thresholds, sponsors are encouraged to propose impurity-specific AI limits supported by sound toxicological reasoning. This approach reflects the FDA’s broader goal of balancing patient safety with practical manufacturing realities.
👉 Understand how AI supports nitrosamine prediction.
Key Features
- Primary Guidance: Control of Nitrosamine Impurities in Human Drugs (2023 update)
- AI Calculation Basis: Linearized multistage cancer risk model targeting a 1 in 100,000 lifetime cancer risk
Data Priority:
- Direct rodent carcinogenicity data (Category 1)
- CPCA and QSAR model predictions (Category 2)
- Read-across from structurally similar nitrosamines (Category 3)
This tiered system ensures that the most reliable data are used first, while still allowing interim controls when information is limited.
👉 Read more about NDSRIs in nitrosamine testing.
Example FDA Thresholds
- NDMA: 96 ng/day
- NDEA: 26.5 ng/day
- NEIPA: 26.5 ng/day
| Compound | FDA AI (ng/day) | Data Source | Category |
|---|---|---|---|
| NDMA | 96 | Animal data | 1 |
| NDEA | 26.5 | CPCA model | 2 |
| NMBA | 96 | Read-across | 3 |
Regulatory Implications
The FDA allows alternative AI limits if supported by strong scientific data. Interim limits are acceptable while additional studies are ongoing. Rather than enforcing strict uniformity, the FDA prioritizes transparency, analytical validation, and risk-based decision-making.
👉 Learn about validated LC–MS/MS nitrosamine testing.
2. EMA’s Conservative Model in Nitrosamine AI Limits Comparison
The EMA applies a standardized and precautionary approach, using TD50-based calculations and cumulative exposure principles.
EMA’s framework aims to reduce variability by enforcing consistent methodologies across products and regions. When uncertainty exists, the agency applies conservative assumptions to ensure long-term patient protection, especially for chronic therapies.
👉 Explore common nitrosamine impurity sources.
Core Elements
- Reference: EMA/409815/2020 – Questions and Answers on Nitrosamine Impurities
- AI Basis: TD50 values from the Carcinogenic Potency Database
- Cumulative Risk Principle: Combined exposure to multiple nitrosamines must remain below the lowest AI
EMA also uses group AI limits for structurally related nitrosamines when compound-specific data are missing. In some cases, AI values are reduced by up to 50% if metabolic or toxicokinetic pathways are unclear.
👉 Review global regulatory expectations.
Example Adjustments
Although EMA aligns with the FDA on NDMA at 96 ng/day, it often applies stricter limits for secondary amine-derived nitrosamines and complex analogs.
| Parameter | EMA Methodology | Impact |
|---|---|---|
| AI Basis | TD50 calculation | Standardized limits |
| Multi-nitrosamine handling | Cumulative exposure | Increased conservatism |
| Interim limits | Allowed for 2 years | Temporary compliance |
| Lifecycle control | Continuous trending | Ongoing verification |
Regulatory Implications
EMA requires detailed process mapping to identify all potential nitrosamine formation pathways. Manufacturers must also perform batch-level trending and meet strict reassessment deadlines as new impurities are identified globally.
👉 Understand the consequences of nitrosamine detection.
3. Health Canada’s Hybrid Approach in Nitrosamine AI Limits Comparison
Health Canada combines EMA’s conservative principles with FDA-like flexibility and strong post-market oversight.
Health Canada largely follows EMA’s TD50-based methodology but allows alternative CPCA or QSAR justifications when scientifically sound. This hybrid approach reflects a balance between patient safety and real-world implementation.
Regulatory Highlights
- Guideline: Control of Nitrosamine Impurities in Human Drugs (Revised March 2023)
- Adopts EMA-style conservative AI derivation
- Allows scientifically justified deviations
- Requires lot-release testing for high-risk products
| Nitrosamine | Health Canada AI (ng/day) | EMA Comparison | Comments |
|---|---|---|---|
| NDMA | 96 | Same | Default value |
| NDEA | 26.5 | Same | EMA-aligned |
| NMPA | 26.5 | Slightly higher | Read-across flexibility |
Compliance Requirements
Health Canada emphasizes CAPA documentation, root cause analysis, and supplier oversight. The agency also encourages predictive toxicology tools, including machine-learning models, signaling early adoption of digital regulatory science.
👉 Learn more about nitrosamine testing in Canada.
4. Comparative Table: Nitrosamine AI Limits Comparison – FDA vs EMA vs Health Canada
| Agency | Methodology Basis | Data Priority | Multi-Nitrosamine Handling | Flexibility | Key Feature |
|---|---|---|---|---|---|
| FDA | CPCA / ICH M7 | Structure–activity | Individual | High | Scientific justification |
| EMA | TD50 | Carcinogenic potency | Cumulative | Moderate | Conservative consistency |
| Health Canada | Hybrid | Read-across | Compound + batch trend | Moderate–High | Post-market control |
5. Impact of Regulatory Divergence on Global Manufacturers
Differences revealed by the Nitrosamine AI limits Comparison create significant regulatory and operational challenges for global manufacturers.
Companies often need separate justifications, testing strategies, and documentation for each region. This increases both workload and compliance risk.
👉 Explore CRO support for nitrosamine risk evaluation.
Key Challenges
- Multiple AI Justifications: Different limits for the same impurity
- Analytical Burden: Varying detection limits and testing frequency
- Labeling Impact: Region-specific updates when limits change
- Risk Management: Continuous global monitoring and reconciliation
Harmonization Outlook
Upcoming ICH M7 (R2) updates and ICH Q13 implementation aim to improve global alignment. AI-driven toxicology tools, including advanced QSAR and CPCA models, are expected to support more consistent potency categorization worldwide. ResolveMass Laboratories Inc. emphasizes cross-agency validation strategies to maintain global compliance.
6. Toward Harmonization: AI-Assisted Toxicology and Nitrosamine AI Limits Comparison
Artificial intelligence is playing a growing role in harmonizing nitrosamine risk assessment.
AI-based toxicology tools can rapidly predict genotoxic potency for new nitrosamines, reducing reliance on long-term animal studies. Shared databases improve transparency, while dynamic models allow AI limits to be updated as new evidence emerges.
Industry Perspective
AI-driven toxicology is becoming both a compliance requirement and a strategic advantage. Health Canada’s acceptance of AI-derived QSAR assessments highlights growing regulatory confidence. Over time, these technologies are expected to support global convergence of nitrosamine AI limits.
Conclusion
The Nitrosamine AI limits Comparison highlights clear regulatory differences between the FDA, EMA, and Health Canada. The FDA favors case-specific scientific flexibility, EMA enforces standardized conservatism, and Health Canada follows a balanced hybrid model. As ICH initiatives advance and AI-based toxicology matures, greater global alignment is likely, supporting a more consistent framework for managing nitrosamine risk.
For expert guidance or tailored compliance strategies, connect with ResolveMass Laboratories Inc.:
👉 Contact Us
FAQs on Nitrosamine AI Limits Comparison
The differences come from how each authority evaluates cancer risk and uncertainty. The FDA focuses on structure-based scientific justification, EMA applies conservative default models, and Health Canada blends both approaches. These philosophies lead to different acceptable intake values for the same impurity.
EMA is generally considered the most conservative regulator in nitrosamine control. It applies cumulative exposure rules and additional safety factors when data are limited. This approach is designed to minimize long-term patient risk, especially for chronic treatments.
Yes, manufacturers can propose alternative AI limits to the FDA and Health Canada. This is allowed when the proposal is supported by strong toxicological data and scientific reasoning. EMA may also accept interim limits but usually within a stricter framework.
CPCA is a structure-based method under ICH M7 that estimates carcinogenic risk. It groups nitrosamines by chemical similarity and predicts potency when animal data are unavailable. This helps regulators set AI limits more efficiently.
TD50 refers to the dose that causes tumors in half of test animals during studies. Regulators use this value to estimate a safe human exposure level. EMA relies heavily on this method for setting conservative AI limits.
Yes, all three agencies allow interim AI limits when complete toxicological data are not yet available. These limits act as temporary controls while further studies are conducted. Manufacturers must still show ongoing efforts to reduce risk.
Reference
- Kruhlak, N. L., Schmidt, M., Froetschl, R., Graber, S., Haas, B., Horne, I., Horne, S., King, S. T., Koval, I. A., Kumaran, G., Langenkamp, A., McGovern, T. J., Peryea, T., Sanh, A., Siqueira Ferreira, A., van Aerts, L., Vespa, A., & Whomsley, R. (2024). Determining recommended acceptable intake limits for N-nitrosamine impurities in pharmaceuticals: Development and application of the Carcinogenic Potency Categorization Approach (CPCA). Regulatory Toxicology and Pharmacology, 150, 105640. https://doi.org/10.1016/j.yrtph.2024.105640
- U.S. Food and Drug Administration. (2023). Recommended acceptable intake limits for nitrosamine drug substance-related impurities (NDSRIs). FDA. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cder-nitrosamine-impurity-acceptable-intake-limits
- Zhang, S., Cheung, J., Kostal, J., Voutchkova-Kostal, A., & Schuler, M. (2025). Re-evaluating acceptable intake: A comparative study of N-nitrosomorpholine and N-nitroso reboxetine potency. Environmental and Molecular Mutagenesis, 66(3), 80–98. https://doi.org/10.1002/em.70007
- Medicines for Europe. (2023, October 17). Review of nitrosamine drug‑substance related impurities (NDSRIs) in pharmaceutical drugs: Risk assessments, acceptable intakes, and QSAR tools (Report). https://www.medicinesforeurope.com/wp-content/uploads/2023/10/Review-of-Nitrosamine-drug-substance-related-impurities-in-Pharma-report.pdf
- European Medicines Agency. (2020). Questions and answers for marketing‑authorisation holders and applicants on nitrosamine impurities in human medicinal products: CHMP opinion (Article 53, Regulation (EC) No 726/2004 referral). https://www.ema.europa.eu/en/documents/opinion-any-scientific-matter/nitrosamines-emea-h-a53-1490-questions-answers-marketing-authorisation-holders-applicants-chmp-opinion-article-53-regulation-ec-no-726-2004-referral-nitrosamine-impurities-human-medicinal-products_en.pdf

