
Introduction: Why Nitrosamines Require a Distinct Focus Within ICH M7 Genotoxic Impurity Testing
When ICH M7 was finalized in 2014, it established a globally accepted scientific framework for Genotoxic Impurity Testing ICH M7 Nitrosamines and other mutagenic impurities present in pharmaceutical drug substances and drug products. However, the nitrosamine crisis that emerged between 2018 and 2019 — initiated by the discovery of NDMA contamination in sartan APIs and later identified in ranitidine and metformin products — revealed a significant operational limitation within the existing framework. Although ICH M7 remained scientifically sound, it was not designed to address the scale, urgency, and technical complexity associated with nitrosamine risk assessment and control.
As a result, the current regulatory environment treats nitrosamines as impurities that technically fall within the ICH M7 framework while simultaneously being governed through far more detailed and specialized regulatory expectations. These expectations are driven by FDA guidance documents, EMA Article 31 referral outcomes, and Health Canada regulatory directives. Understanding how nitrosamines align with the structure of ICH M7 — and where they diverge from conventional M7 impurity management practices — has become essential for pharmaceutical manufacturers, analytical laboratories, and regulatory affairs teams responsible for ensuring product quality and compliance.
Access Comprehensive Impurity Analysis: Learn how to systematically identify and mitigate complex genotoxic risks in your pharmaceutical pipeline. Explore our Nitrosamine Analysis Services.
Share via:
Article Summary:
- Nitrosamines require specialized handling within the ICH M7 framework because the original guideline was not designed to manage the scale, urgency, and complexity of nitrosamine contamination discovered in drugs like sartans, ranitidine, and metformin.
- Most nitrosamines fall into ICH M7 Class 1 or Class 2 impurities, but their carcinogenic potency varies widely, making compound-specific acceptable intake (AI) limits more important than classification alone.
- (Q)SAR assessment works differently for nitrosamines because the N-nitroso functional group is highly predictive of mutagenicity, often reducing the need for confirmatory Ames testing.
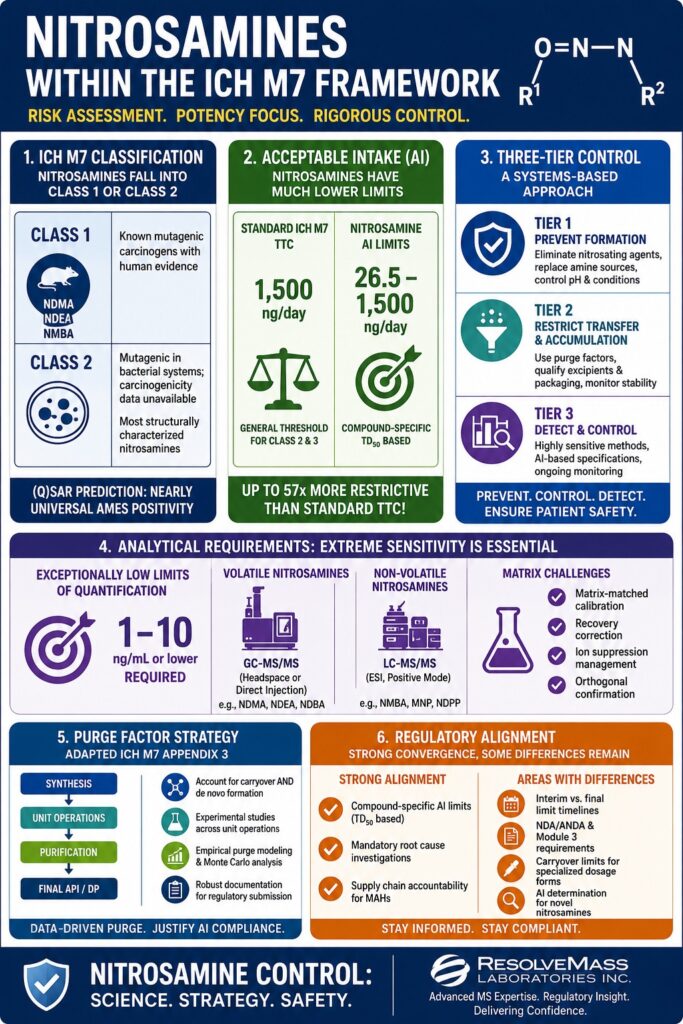
- Nitrosamine AI limits are far stricter than standard ICH M7 TTC limits, with some compounds like NDMA limited to 26.5 ng/day — up to 57 times more restrictive than traditional thresholds.
- Nitrosamine risk management requires a systems-based control strategy, involving synthetic chemistry, formulation development, packaging, toxicology, analytical science, and stability studies working together.
- The recommended three-tier control strategy includes:
- Preventing nitrosamine formation
- Restricting transfer and accumulation
- Detecting contamination using ultra-sensitive analytical methods
- Nitrosamine testing demands highly sensitive analytical technologies such as LC-MS/MS, GC-MS/MS, and HRMS, often requiring detection limits as low as 1–10 ng/mL in complex pharmaceutical matrices.
- Global regulators like the FDA, EMA, and Health Canada largely agree on nitrosamine risk principles, but differences still exist in implementation timelines, AI determination methods, and submission expectations for pharmaceutical companies.
How the ICH M7 Classification Framework Applies — and Becomes Challenging — for Nitrosamines
ICH M7 categorizes mutagenic impurities into five separate classes. Nitrosamines almost always fall into either Class 1 or Class 2. However, because nitrosamines share highly consistent structural and biological characteristics, the classification process itself often provides less practical differentiation compared to other impurity categories evaluated under ICH M7.
ICH M7 Classification Schema and Nitrosamine Alignment
| Class | Definition | Nitrosamine Relevance |
|---|---|---|
| Class 1 | Known mutagenic carcinogens with human evidence | NDMA, NDEA, and NMBA are established rodent carcinogens with strong CoC designation |
| Class 2 | Mutagenic in bacterial systems; carcinogenicity data unavailable | Most structurally characterized nitrosamines with positive Ames results |
| Class 3 | Structural alert present, but no Ames or carcinogenicity data available | Rarely applicable because most nitrosamines demonstrate Ames positivity |
| Class 4 | Structural alert with demonstrated non-mutagenicity | Generally not applicable to N-nitrosamines |
| Class 5 | No structural alert or sufficient negative data | Explicitly not applicable to nitrosamines |
The elegance of the ICH M7 classification system lies in its ability to use impurity class assignments to establish toxicological control strategies and acceptable exposure limits. However, this framework becomes less efficient when applied to nitrosamines because the majority of nitrosamines consistently fall into the same classifications, while their carcinogenic potency can vary dramatically across several orders of magnitude.
For example:
- NDMA has an acceptable intake (AI) limit of 26.5 ng/day
- NMBA has an AI of 96 ng/day
- Certain lower-potency nitrosamines may receive interim limits approaching 1,500 ng/day under FDA potency-based approaches
This wide potency variation demonstrates why compound-specific PDEs and AI values — derived from TD50 data and CPDB analyses — have become the primary operational basis for nitrosamine risk management rather than M7 classification alone.
Evaluate High-Risk Formulations: Different drug classes carry unique risks. Review targeted screening strategies by reading our guide on Nitrosamine Testing for High-Risk Drug Classes.
The (Q)SAR Paradox: Why In Silico Assessment Functions Differently for Nitrosamines
(Q)SAR methodologies are central to standard ICH M7 implementation. However, for nitrosamines, their role becomes unusually straightforward — not because the models fail, but because they perform with remarkable consistency.
Under traditional ICH M7 workflows, the process generally follows these steps:
- Identify a structural alert using (Q)SAR tools
- Evaluate the relevance of the alert
- Confirm or refute mutagenicity using Ames testing
- Assign the impurity classification
- Establish toxicological limits
For most impurity classes, this process creates meaningful differentiation because many structural alerts may be context-dependent or generate false-positive signals. Nitrosamines behave differently. The N-nitroso functional group is among the most reliably predictive mutagenic structural alerts in modern toxicological assessment.
Published literature demonstrates that the Ames positivity rate for N-nitrosamines exceeds approximately 85–90%, while smaller and more volatile nitrosamines such as NDMA and DEA-derived analogs approach nearly universal positivity rates.
This creates two major operational consequences:
1. Reduced Reliance on Confirmatory Ames Testing
Regulatory agencies increasingly accept the omission of confirmatory Ames testing for structurally obvious nitrosamines because the (Q)SAR prediction itself is considered sufficient to support a Class 2 assignment. This significantly accelerates risk assessment timelines.
2. Greater Emphasis on Potency Assessment
Once mutagenic classification is effectively assumed, the primary regulatory focus shifts toward carcinogenic potency determination. The critical parameter becomes the TD50-derived acceptable intake rather than the binary question of whether mutagenicity exists.
As a result, databases such as the Carcinogenic Potency Database (CPDB) and FDA nitrosamine AI reference tables have become more operationally important than the (Q)SAR systems themselves during nitrosamine evaluations.
This represents a meaningful departure from conventional ICH M7 implementation, where the sequence of (Q)SAR assessment, Ames testing, and classification contains multiple scientifically significant decision points.
Stay Current with Evolving Guidance: Understand how recent framework revisions affect your structural alerts and read across strategies. Read our insight on the Impact of ICH M7(R2) Updates on Nitrosamine Risk Assessment.
Acceptable Intake Limits: Why Nitrosamine PDEs Differ Fundamentally from Standard M7 TTC Approaches
Traditional ICH M7 methodology applies a Threshold of Toxicological Concern (TTC) of 1.5 µg/day for most Class 2 and Class 3 impurities. Nitrosamines classified within the cohort of concern (CoC), however, are subject to dramatically lower acceptable intake limits ranging from approximately 26.5 ng/day to 1,500 ng/day.
The TTC principle in ICH M7 was originally developed using a linear extrapolation model corresponding to a theoretical excess cancer risk of 1 in 100,000 over a lifetime exposure at 1.5 µg/day. While this approach functions effectively as a conservative general threshold, the ICH M7 guideline explicitly excludes N-nitroso compounds from TTC applicability. Instead, compound-specific risk assessments are mandatory.
Comparison of Standard TTC Versus Nitrosamine-Specific AI Limits
| Compound | Standard ICH M7 TTC | Compound-Specific AI | Relative Restrictiveness |
|---|---|---|---|
| NDMA | 1,500 ng/day | 26.5 ng/day | ~57x more restrictive |
| NDEA | 1,500 ng/day | 26.5 ng/day | ~57x more restrictive |
| NMBA | 1,500 ng/day | 96 ng/day | ~16x more restrictive |
| NDIPA | 1,500 ng/day | 26.5 ng/day | ~57x more restrictive |
| NMPA | 1,500 ng/day | 96 ng/day | ~16x more restrictive |
These differences are operationally significant because they directly determine specification limits and required analytical method sensitivity. Analytical methods originally validated for conventional M7 impurities at 1.5 µg/day sensitivity may fail to detect nitrosamine contamination levels by factors exceeding 50-fold.
Compare International AI Standards: Limits and classifications vary slightly by jurisdiction. View our global matrix on Nitrosamine AI Limits Comparison.
Root Cause Investigation and Control Strategy: A Fundamentally Different Operational Model
Traditional non-nitrosamine M7 impurity programs primarily focus on synthetic route optimization and specification management. Nitrosamine control strategies require a far broader and more integrated approach centered on root cause investigation and formation pathway analysis.
Regulatory agencies including the FDA, EMA, and Health Canada require manufacturers to perform comprehensive nitrosamine risk assessments addressing:
- Potential nitrosating agents present during synthesis, formulation, or packaging
- Secondary amine sources originating from APIs, degradants, excipients, or reagents
- Drug product-level nitrosamine formation during shelf life
- Nitrite-containing excipients capable of facilitating nitrosation
- Packaging and storage conditions contributing to nitrosamine generation
- Nitrocellulose-containing packaging materials capable of introducing contamination
This differs substantially from conventional ICH M7 control strategies, which generally focus on eliminating a mutagenic reagent or controlling a known synthetic impurity through specification limits.
Nitrosamine risk management instead requires a systems-based strategy involving:
- Synthetic chemistry
- Formulation development
- Packaging engineering
- Analytical chemistry
- Toxicology
- Stability science
all functioning simultaneously within a coordinated control program.
The Three-Tier Nitrosamine Control Strategy
Tier 1 — Prevention of Formation
- Remove nitrosating agents from manufacturing processes
- Replace secondary amine-containing reagents whenever possible
- Modify pH conditions and reaction environments to minimize nitrosation potential
Tier 2 — Restriction of Transfer and Accumulation
- Apply validated purge factor calculations using adapted ICH M7 Appendix 3 methodologies
- Qualify excipients and packaging materials for nitrite content and contamination risk
- Conduct stability studies focused specifically on nitrosamine formation potential
Tier 3 — Detection and Analytical Control
- Implement highly sensitive analytical technologies such as LC-MS/MS, GC-MS/MS, and GC-TEA
- Establish specification limits at or below compound-specific AI thresholds
- Validate analytical methods according to nitrosamine-focused FDA and EMA expectations rather than relying solely on traditional ICH Q2(R1) validation standards
Evaluate Packaging Risks: Don’t let your packaging compromise compliance. Read our technical breakdown on Packaging Leachables and Nitrosamine E&L Studies.
Analytical Method Requirements: Sub-PPM Sensitivity Is Essential
Nitrosamine analysis within the ICH M7 framework requires exceptionally low limits of quantification, often reaching 1–10 ng/mL or lower within highly complex pharmaceutical matrices. This level of sensitivity requires specialized instrumentation and analytical expertise that extends well beyond conventional impurity testing programs.
The analytical challenges associated with nitrosamines are particularly demanding:
Volatile Nitrosamines
Compounds such as NDMA, NDEA, and NDBA are commonly analyzed using GC-MS/MS systems with headspace sampling or direct injection methodologies. The FDA has also published GC-HRMS methods that serve as reference approaches.
Non-Volatile Nitrosamines
Compounds including NMBA, MNP, and NDPP typically require LC-MS/MS analysis utilizing electrospray ionization and highly specific positive-mode MRM transitions.
Ranitidine-Related Nitrosamines
NDMA formation associated with ranitidine degradation required orthogonal analytical methodologies due to in-source fragmentation artifacts capable of generating false-positive signals.
Matrix Interference Challenges
Drug product matrices such as tablets, capsules, and injectables frequently introduce co-elution effects and ion suppression complications. These issues necessitate:
- Matrix-matched calibration
- Recovery correction studies
- Rigorous specificity validation
- Orthogonal confirmation approaches
FDA analytical method repositories and EMA reflection papers have effectively become the dominant reference standards for nitrosamine method validation, surpassing traditional ICH Q2(R1) expectations in practical regulatory importance.
Minimum Analytical Performance Expectations
| Parameter | Standard M7 Method | Nitrosamine-Specific Requirement |
|---|---|---|
| LOQ | Approximately 0.05–0.1% of API | Approximately 0.03–0.3 ppm in API or drug product |
| Matrix validation | ICH Q2(R1) requirements | Agency-specific plus matrix-matched recovery validation |
| Reference standards | USP/EP compendial standards | Certified nitrosamine reference materials, preferably isotopically labeled |
| Instrumentation | HPLC-UV often sufficient | MS/MS or HRMS considered mandatory |
| Specificity confirmation | Chromatographic retention | MRM transition confirmation, retention time, and isotope ratio evaluation |
Achieve Ultra-Low Quantification: When standard sensitivity falls short, specialized instrumentation is required. Learn about our Ultra-Low Limit of Quantitation (LOQ) in Nitrosamine Testing.
Purge Factor Strategy for Nitrosamines: Applying ICH M7 Appendix 3 With Modifications
Purge factor calculations described in ICH M7 Appendix 3 can be adapted for nitrosamine control strategies, although the standard assumptions frequently require substantial modification.
Appendix 3 was originally developed to estimate the removal of synthetic impurities throughout manufacturing operations. Nitrosamines introduce two major complications into this model:
1. Nitrosamine Formation May Occur Throughout the Process
Unlike conventional synthetic impurities introduced at an early manufacturing stage and progressively purged, nitrosamines can form at multiple process stages. Therefore, purge calculations must evaluate both carryover and de novo formation risk.
2. Default Purge Assumptions Are Often Excessively Conservative
The original Appendix 3 factors were calibrated primarily for solid reagent impurities. Highly volatile nitrosamines such as NDMA and NDEA often purge much more efficiently during:
- Distillation
- Aqueous washing
- Drying operations
- Solvent exchange processes
This means experimentally validated purge studies can frequently justify higher process limits while remaining compliant with final AI requirements.
Successful nitrosamine purge programs often combine:
- Experimental partition coefficient determination
- Spiking studies across unit operations
- Empirical purge modeling
- Monte Carlo simulations for variability assessment
- Documentation packages designed for regulatory submission and inspection readiness
Optimize Your Control Strategy: Quantify the reduction of risks during manufacturing processing. Learn more about Nitrosamine Purge Factor Calculation and Validation.
Regulatory Convergence and Continuing Global Differences
The FDA, EMA, Health Canada, and ICH have achieved substantial alignment regarding the fundamental principles of nitrosamine risk assessment. Nevertheless, important jurisdiction-specific differences remain.
Areas of Strong Regulatory Alignment
- Acceptance of compound-specific AI limits derived from TD50 linear extrapolation models
- Mandatory root cause investigations for all relevant products
- Supply chain accountability assigned to marketing authorization holders
Areas Where Differences Persist
Interim Versus Final Limits
The FDA has issued multiple implementation deadline extensions for certain product categories, whereas EMA timelines have generally remained more rigid.
Requirements for New Marketing Applications
FDA submissions require formal nitrosamine risk assessments within NDA and ANDA filings. EMA expectations continue evolving within Module 3 submission requirements.
Carryover Expectations for Specialized Dosage Forms
Acceptable nitrosamine limits for inhalation products and topical formulations remain under active regulatory evaluation.
AI Determination for Novel Nitrosamines
When direct CPDB carcinogenicity data are unavailable, regulatory agencies may differ regarding acceptable read-across methodologies and structure-activity relationship approaches used to derive provisional AI values.
Streamline Your Generic Drug Approvals: Ensure your regulatory filings meet strict requirements. See our approach to Nitrosamine Risk Assessment for ANDA Submission.
Conclusion: Nitrosamines Represent the Most Demanding Application of ICH M7 Genotoxic Impurity Testing
The ICH M7 framework remains the scientific foundation for modern genotoxic impurity assessment. However, nitrosamines have evolved into a uniquely demanding category requiring substantially more rigorous analytical, toxicological, and regulatory control strategies than traditional mutagenic impurities.
The combination of cohort-of-concern designation, near-universal Ames positivity, nanogram-level acceptable intake requirements, and the challenge of managing formation-based contamination rather than simple carryover has effectively created a specialized nitrosamine regulatory discipline operating within the broader ICH M7 architecture.
Partner with Testing Experts: Ready to develop robust, ultra-sensitive assays for your complex matrices? Reach out for GC-MS Method Development for Nitrosamine Testing.
For organizations responsible for Genotoxic Impurity Testing ICH M7 Nitrosamines compliance, long-term success depends on three essential capabilities:
- Understanding precisely how nitrosamine risks intersect with M7 classification and cohort-of-concern principles
- Implementing analytical methodologies capable of achieving the sensitivity and selectivity required by compound-specific AI limits
- Developing root-cause-driven control strategies that extend beyond traditional specification management approaches
ResolveMass Laboratories Inc. combines advanced high-resolution mass spectrometry expertise, extensive ICH M7 regulatory knowledge, and practical experience in nitrosamine method development and validation — precisely where analytical science and regulatory strategy must operate together to achieve compliance and product safety.
Frequently Asked Questions (FAQs)
Nitrosamines are not automatically assigned to Class 1 or Class 2, but most of them ultimately fall into these categories because of their well-established mutagenic behavior. The N-nitroso functional group is recognized as a highly reliable structural alert in toxicological evaluation, and many nitrosamines consistently produce positive Ames test results. Class 1 classification generally applies when carcinogenicity data in humans or animals is available, while Class 2 is used when mutagenicity is confirmed without complete carcinogenicity data.
No, the standard Threshold of Toxicological Concern (TTC) under ICH M7 does not apply to nitrosamines. N-nitroso compounds are specifically included within the cohort of concern, which requires compound-specific acceptable intake limits instead of the generic TTC approach. As a result, highly potent nitrosamines such as NDMA and NDEA are controlled at extremely low levels, often in the nanogram-per-day range.
For nitrosamines, purge factor calculations must evaluate both impurity removal and the possibility of new nitrosamine formation during processing. Traditional ICH M7 calculations assume the impurity enters the process at a fixed level and gradually decreases through manufacturing steps. Nitrosamines behave differently because they may form at multiple stages if nitrosating conditions are present. Therefore, manufacturers are expected to support purge claims with experimental studies and process-specific validation data.
In nitrosamine guidance documents, AI and PDE are often used interchangeably because both represent toxicological exposure limits intended to protect patient safety. Technically, AI is the terminology commonly used within ICH M7, while PDE is more frequently associated with ICH Q3C guidance for residual solvents. Despite the terminology difference, both values are generally derived using similar carcinogenic risk assessment principles based on TD50 data and lifetime cancer risk calculations.
GC-MS/MS is widely preferred for volatile nitrosamines such as NDMA, NDEA, and NDBA because it provides excellent sensitivity and selectivity for low-level detection. However, non-volatile or larger nitrosamine structures are more effectively analyzed using LC-MS/MS techniques. The selection of analytical instrumentation depends on the physical and chemical characteristics of the target nitrosamine, as well as the complexity of the sample matrix.
When direct carcinogenicity data is unavailable, regulatory agencies generally allow the use of read-across approaches based on structurally similar nitrosamines with established toxicological profiles. The selected analogue should represent a conservative comparison, often using the most potent related compound as the reference point. Manufacturers must provide scientific justification explaining structural similarity, uncertainty considerations, and the rationale behind the proposed acceptable intake limit.
Isotopically labeled internal standards play a critical role in improving the accuracy and reliability of nitrosamine quantification by MS/MS and HRMS methods. These standards help compensate for matrix-related ionization effects, variations in sample preparation, and changes in chromatographic recovery. Their use ensures more consistent analytical performance across complex pharmaceutical matrices and is increasingly viewed as an essential component of robust nitrosamine method validation.
Reference:
- López Rodríguez, R., McManus, J. A., Murphy, N. S., Ott, M. A., & Burns, M. J. (2020). Pathways for N-nitroso compound formation: Secondary amines and beyond. Organic Process Research & Development, 24(9), 1558–1585. https://doi.org/10.1021/acs.oprd.0c00323
- U.S. Food and Drug Administration. (2024). Control of nitrosamine impurities in human drugs: Guidance for industry (Revision 2). U.S. Department of Health and Human Services. https://www.fda.gov/media/141720/download
- Cioc, R. C., Joyce, C., Mayr, M., & Bream, R. N. (2023). Formation of N-nitrosamine drug substance related impurities in medicines: A regulatory perspective on risk factors and mitigation strategies. Organic Process Research & Development, 27(10), 1736–1750. https://doi.org/10.1021/acs.oprd.3c00153

