
Introduction
Within the increasingly competitive biopharmaceutical industry, performing a comprehensive Aggregation Analysis in Biosimilars is essential for establishing analytical similarity and safeguarding patient health. Protein aggregation remains one of the most significant challenges associated with biotherapeutic development, as it can directly influence product efficacy, stability, shelf life, and immunogenicity. For biosimilar manufacturers, demonstrating that the aggregate profile of a biosimilar closely aligns with that of the reference biologic is a critical regulatory requirement.
To ensure success, developers must align their analytical strategy with established Critical Quality Attributes (CQAs) in Biosimilars.
Traditionally, protein characterization relied on biophysical techniques that offered limited resolution or generated averaged measurements across populations of molecules. However, modern high-resolution mass spectrometry (MS) has become a leading analytical platform for identifying, characterizing, and structurally resolving complex protein aggregates. Its ability to provide detailed molecular insights makes it indispensable for contemporary biosimilar development programs.
Share via:
Article Summary:
- Aggregation analysis is a critical part of biosimilar development, as protein aggregates can impact product safety, efficacy, stability, and immunogenicity. Regulatory agencies require biosimilars to demonstrate aggregate profiles comparable to their reference products.
- Global regulatory frameworks, including FDA and EMA guidelines, emphasize analytical comparability as a key component of biosimilar approval. Strong analytical evidence can reduce the need for extensive clinical studies under the totality-of-evidence approach.
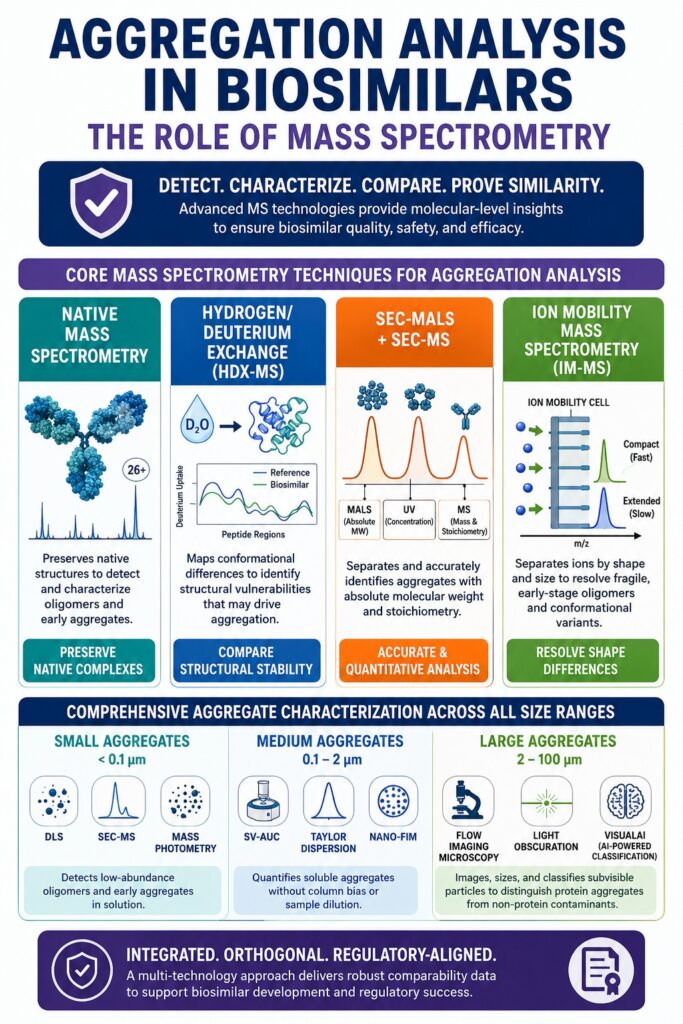
- Mass spectrometry (MS) has become the gold standard for aggregation analysis, providing detailed molecular, structural, and stoichiometric information that traditional techniques such as DLS and SEC-UV may miss.
- Native mass spectrometry enables detection of low-abundance protein aggregates while preserving their natural structure, making it highly effective for identifying early aggregation events, oligomers, and non-covalent protein complexes.
- Hydrogen/Deuterium Exchange Mass Spectrometry (HDX-MS) helps assess higher-order structural similarity by identifying conformational differences and aggregation-prone regions between biosimilars and reference products.
- SEC-MS, SEC-MALS, and Ion Mobility Mass Spectrometry (IM-MS) provide complementary insights into aggregate size, molecular weight, shape, and composition, improving the accuracy of aggregate characterization.
- A robust biosimilar aggregation strategy combines advanced mass spectrometry with orthogonal techniques such as SV-AUC, Flow Imaging Microscopy, and particle analysis methods to generate the comprehensive data package required for regulatory approval and biosimilar success.
Regulatory Frameworks for Aggregation Analysis in Biosimilars
Comprehensive aggregate characterization is required by regulatory agencies worldwide because protein aggregates are considered critical immunogenic impurities that may adversely affect both safety and therapeutic performance. Demonstrating a highly comparable aggregate profile relative to the reference product is a central component of the evidence package needed for biosimilar approval.
Under the established “totality of evidence” approach, developers can potentially reduce or eliminate the need for extensive clinical efficacy studies by generating robust analytical comparability data. Protein aggregates present in parenteral formulations have the potential to trigger severe anti-drug antibody (ADA) responses, making their characterization and control a major regulatory concern. Consequently, guidelines such as ICH Q6B classify aggregates as critical quality-related impurities that require stringent monitoring and control.
The foundation for these regulatory filings is built upon rigorous Biosimilar Comparability Studies.
Recent regulatory developments have further strengthened the importance of analytical characterization. Draft guidance documents released by the FDA in late 2025 and finalized in March 2026 emphasized that Comparative Analytical Assessment (CAA) can often detect subtle but clinically meaningful differences with greater sensitivity than traditional comparative clinical efficacy studies. This evolving regulatory perspective places advanced structural characterization and molecular fingerprinting at the forefront of biosimilar development.
In both the United States and the European Union, detailed analytical comparisons between biosimilars and their corresponding reference products are mandatory. In the United States, this requirement is fulfilled through the 351(k) pathway established under the Biologics Price Competition and Innovation Act (BPCIA). In the European Union, biosimilar applications are submitted under Article 10(4) of Directive 2001/83/EC. Regulatory submissions must demonstrate aggregate comparability across multiple manufacturing lots to confirm that the biosimilar remains within the natural variability range of the innovator product.
The growing complexity of biosimilar submissions is reflected in historical approval trends. Between 2016 and 2020, the FDA approved 93 Biologics License Applications (BLAs) through accelerated electronic review pathways, with biosimilars accounting for approximately 30% of those approvals. Monoclonal antibodies (mAbs) represented 71% of approved biosimilars, highlighting the increasing demand for sophisticated analytical approaches capable of characterizing highly complex protein therapeutics.
Regulatory Comparison
| Regulatory Parameter | FDA (United States – 351(k) Pathway) | EMA (European Union – Article 10(4)) |
|---|---|---|
| Primary Approval Framework | Totality of Evidence (stepwise, risk-based) | Totality of Evidence (stepwise, guideline-driven) |
| Analytical Comparability | Foundational; analytical fingerprinting strongly encouraged | Mandatory; advanced characterization expected |
| Non-Clinical In Vivo Studies | Frequently waived with strong analytical evidence | Preference for minimal or no animal studies |
| Efficacy Trial Requirements | May be waived when PK/PD and analytical similarity are highly convincing | Confirmatory studies may be expected for highly complex molecules |
| Comparator Sourcing | March 2026 revisions permit scientifically justified non-US comparators without mandatory three-way bridging | Community-approved reference products accepted with rigorous comparability exercises |
| Interchangeability | Requires dedicated switching studies for pharmacy-level substitution | Managed as a substitution policy by individual member states |
Explore our specialized Impurity Profiling of Biosimilars services.
Core Mass Spectrometry Methodologies for Aggregation Analysis in Biosimilars
Mass spectrometry is widely regarded as the gold-standard analytical technology for aggregate characterization because it provides precise molecular weight measurements, stoichiometric information, and structural insights into individual species present within heterogeneous protein mixtures. This capability overcomes many of the limitations associated with conventional ensemble-averaging analytical techniques.
Protein aggregation is driven by a complex thermodynamic landscape in which partially unfolded or misfolded monomeric intermediates self-associate into soluble oligomers that may subsequently evolve into larger insoluble aggregates and fibrillar structures. Traditional techniques such as Dynamic Light Scattering (DLS) and Size-Exclusion Chromatography with UV detection (SEC-UV) generally provide averaged measurements and may fail to detect low-abundance yet highly immunogenic aggregate populations.
To handle complex mixtures, Intact Mass Analysis for Biosimilars is the starting point for confirming identity and purity.
Mass spectrometry, particularly when coupled with electrospray ionization (ESI), enables direct transfer of solution-phase protein complexes into the gas phase while preserving key structural features. This allows detailed examination of oligomeric distributions and aggregate architectures that would otherwise remain unresolved.
Native Mass Spectrometry in Aggregation Analysis in Biosimilars
Native mass spectrometry preserves non-covalent interactions and quaternary protein structures by introducing biomolecules into the gas phase directly from physiologically relevant volatile buffers. This approach facilitates highly sensitive detection of low-abundance oligomeric intermediates while maintaining native-like conformations.
Native Mass Spectrometry for Biosimilars provides the necessary resolution to distinguish between native and non-native aggregate states.
Under conventional denaturing MS conditions involving acidic environments and organic solvents, proteins unfold extensively and produce highly charged ions that obscure information regarding higher-order structure. In contrast, native MS maintains proteins in a folded state, reducing charge accumulation and preserving biologically relevant structural assemblies.
As a result, native mass spectra typically display narrower charge-state distributions. For example, intact IgG monoclonal antibodies commonly exhibit charge states ranging from +26 to +30 under native conditions, compared with approximately +20 to +70 under denaturing conditions. These reduced charge states shift signals toward higher mass-to-charge ratios (m/z > 5,000), significantly improving spectral clarity and separation.
Advanced high-mass detection platforms, including the SCIEX ZenoTOF 8600 and Orbitrap systems equipped with dedicated biopharmaceutical workflows, enable characterization of non-covalent complexes exceeding 800 kDa while requiring only minimal sample quantities.
Native MS Characteristics
- High Charge States (+20 to +70) → Low m/z (2,000 to 4,000) → Unfolded proteins with disrupted non-covalent interactions
- Low Charge States (+26 to +30) → High m/z (> 5,000) → Preserved native aggregate architectures
During forced degradation and comparability assessments, native MS can identify early aggregation events, including protein misfolding and dimer formation. The technique can distinguish between native dimers and partially unfolded aggregation-prone intermediates through analysis of charge-state distributions.
Additionally, optimized low-energy source conditions on platforms such as the SCIEX ZenoTOF 8600 minimize in-source fragmentation, enabling successful detection of low-abundance dimers in stressed trastuzumab formulations. Similar methodologies have been used to characterize fragile cysteine-linked antibody-drug conjugates (ADCs), including trastuzumab-deruxtecan, while preserving structural integrity.
Review our comprehensive protocols for Forced Degradation of Biosimilars.
Hydrogen/Deuterium Exchange Mass Spectrometry (HDX-MS) and Aggregation Analysis in Biosimilars
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) is a powerful technique used to investigate conformational dynamics and identify structurally vulnerable regions within biosimilars. By measuring the rate at which backbone amide hydrogens exchange with deuterium in solution, HDX-MS reveals areas of structural instability that may initiate aggregation.
Amide hydrogens located within stable secondary structural elements, including β-sheets and α-helices, or buried within hydrophobic cores, exchange significantly more slowly than hydrogens located in flexible solvent-exposed regions. During biosimilar comparability studies, both the biosimilar candidate and the reference product undergo parallel deuterium labeling experiments.
Following labeling, samples are quenched and digested using acid-stable proteases such as pepsin. The resulting peptides are analyzed using rapid low-temperature chromatography coupled to high-resolution mass spectrometry. Bottom-up peptide mapping generated through pepsin digestion provides extensive sequence coverage and allows precise localization of conformational differences.
When a biosimilar possesses an equivalent higher-order structure to the reference product, deuterium uptake profiles produce highly similar mirror plots across all peptide regions. Any structural discrepancy, including localized unfolding or disruption of disulfide bonds, increases solvent accessibility and results in measurable changes in deuterium incorporation.
HDX-MS Workflow
Step 1: Deuterium Labeling
Incubate biosimilar and reference products in D₂O buffer over defined time intervals ranging from 30 seconds to 60 minutes.
Step 2: Quenching
Reduce pH to 2.5 and temperature to 0°C to halt labeling and minimize back-exchange.
Step 3: Digestion
Perform online proteolysis using pepsin or similar acid-stable enzymes to generate region-specific peptides.
Step 4: Rapid LC-MS Analysis
Conduct low-temperature chromatography coupled with high-resolution mass spectrometry.
Step 5: Comparative Structural Mapping
Generate deuterium uptake mirror plots to evaluate higher-order structural equivalence.
Modern HDX-MS platforms employ automated robotic systems, including the LEAP Technologies H/D-X PAL sampler, equipped with temperature-controlled environments that maintain strict 0°C quench conditions and reduce experimental variability.
Researchers at NIST have further advanced HDX-MS by developing subzero-temperature chromatography systems operating at -30°C using specially formulated mobile phases. Extending chromatographic gradients to 40 minutes under these conditions enables recovery of approximately 16% to 26% more retained deuterium compared with conventional 8-minute gradients conducted at 0°C, substantially improving analytical confidence.
To eliminate carryover from highly hydrophobic aggregate species, advanced automated workflows incorporate specialized washing procedures using FOS-choline-12 (n-dodecylphosphocholine).
Online Size-Exclusion Chromatography-Mass Spectrometry (SEC-MS) and SEC-MALS Integration
SEC-MS and SEC-MALS provide quantitative and standard-independent characterization of soluble protein aggregates according to their hydrodynamic properties. Direct coupling of SEC systems with mass spectrometers enables highly accurate identification of co-eluting species and formulation-related impurities.
Conventional SEC methods typically employ high-salt mobile phases and non-volatile phosphate buffers that are incompatible with electrospray ionization. To address this challenge, modern SEC-MS workflows utilize volatile mobile phases such as 50 mM ammonium formate or ammonium acetate, allowing seamless online coupling with mass spectrometric detectors.
SEC separates monomeric proteins from dimers, trimers, and larger aggregate species. As these fractions elute, the mass spectrometer accurately determines molecular masses and stoichiometries, enabling definitive identification of each chromatographic peak. This capability reduces the likelihood of false-positive aggregate assignments caused by co-eluting host cell proteins or non-protein formulation components.
For example, online SEC-MS analysis performed on heat-stressed protein mixtures using a Waters BioSuite 250 column (7.8 mm × 300 mm, 5 μm) successfully separated and identified BSA monomer aggregates with a molecular mass of 66,524 Da, eluting at approximately 5.80 minutes using a 4:1 post-column split ratio directed toward UV and ESI-MS detection.
The addition of multi-angle light scattering (MALS), differential refractive index (dRI), and UV detectors in SEC-UV-MALS-dRI systems provides a calibration-free approach for determining absolute molecular weight throughout the chromatographic profile.
Because larger aggregates scatter light more intensely, SEC-MALS demonstrates exceptional sensitivity toward trace high-molecular-weight species that may be underestimated using UV detection alone. Moreover, SEC-MALS can reveal concentration-dependent self-association behavior and monitor protein recovery across the chromatographic process, helping identify losses caused by adsorption or shear-induced aggregate disruption.
Comparative Analytical Capabilities
| Feature / Capability | SEC-UV | SEC-MALS | SEC-MS |
| Separation Principle | Hydrodynamic Volume | Hydrodynamic Volume | Hydrodynamic Volume |
| Mass Determination | Relative; calibration-dependent | Absolute; standard-free | Absolute; highly precise |
| Buffer Compatibility | Highly tolerant of non-volatile buffers | Highly tolerant of non-volatile buffers | Requires volatile mobile phases |
| Sensitivity to Co-eluting Species | Poor | Moderate | Excellent |
| Modification Identification | No | No | Yes |
| Shear Force / Dilution Effects | High | High | High |
Ion Mobility Mass Spectrometry in Advanced Aggregation Analysis in Biosimilars
Ion mobility mass spectrometry (IM-MS) provides an additional dimension of separation by distinguishing aggregate species according to their three-dimensional shape and collision cross-section. This capability allows characterization of fragile, early-stage oligomers that often remain unresolved by conventional mass spectrometry alone.
Traditional MS separates ions exclusively according to their mass-to-charge ratio. Consequently, distinct conformations or oligomeric assemblies possessing identical m/z values may appear indistinguishable. IM-MS addresses this limitation by introducing gas-phase separation based on molecular shape and size.
As ions travel through a drift region containing inert gases such as nitrogen or helium under an applied electric field, their movement is influenced by overall geometry. Compact conformations encounter fewer collisions and migrate more rapidly than extended structures of identical mass. The resulting measurements are translated into collision cross-section (CCS) values, which provide reproducible descriptors of molecular architecture.
IM-MS Separation Process
Ion Mobility Cell
(Separation Based on Shape and Size)
- Compact conformers migrate rapidly.
- Extended conformers migrate more slowly.
Mass Analyzer
- Separation according to m/z.
Output
- Intensity
- Collision Cross-Section (CCS)
- Mass-to-Charge Ratio (m/z)
For biosimilar aggregate analysis, Trapped Ion Mobility Spectrometry coupled with quadrupole time-of-flight mass spectrometry (TIMS-qToF-MS) offers exceptional resolving power. TIMS retains ions within an RF funnel against a counter-flowing gas stream and releases them selectively according to mobility characteristics.
When operated under carefully optimized low-energy conditions, TIMS preserves fragile non-covalent assemblies and prevents unwanted gas-phase dissociation. This capability enables researchers to characterize heterogeneous conformational ensembles and identify early oligomeric species associated with amyloidogenic or amorphous aggregation pathways.
Studies involving α-synuclein have demonstrated the ability of IM-MS to distinguish among multiple proteoforms, including unmodified monomers, phosphorylated variants, and oxidized species, while revealing how post-translational modifications influence aggregation behavior. Similar applications have been reported for copper/zinc superoxide dismutase (SOD1), where IM-MS has been used to track conformational changes associated with metal loss and subsequent self-assembly processes.
Subvisible Particle Detection and Orthogonal Characterization Frameworks
Comprehensive characterization of subvisible particles ranging from 0.1 to 100 micrometers requires a combination of orthogonal analytical techniques. No single analytical method is capable of accurately characterizing protein aggregates across this entire size spectrum, making integration of mass spectrometry, imaging technologies, and column-free methodologies essential.
Historically, USP chapters focused primarily on particles larger than 10 μm in injectable products. However, extensive research has demonstrated that particles within the 0.1 μm to 10 μm range can be highly immunogenic and capable of stimulating significant immune responses. Consequently, regulatory authorities now expect active monitoring and characterization of these smaller particle populations.
Aggregate Characterization by Size Range
Small Aggregates (<0.1 μm)
- Dynamic Light Scattering (DLS)
- SEC-MS
- Mass Photometry
Medium Aggregates (0.1–2 μm)
- Sedimentation Velocity-Analytical Ultracentrifugation (SV-AUC)
- Taylor Dispersion Analysis
- Nano-FIM
Large Aggregates (2–100 μm)
- Flow Imaging Microscopy (FlowCam)
- Light Obscuration
To overcome limitations associated with SEC, including aggregate loss through filtration, dilution-induced dissociation, and shear-related disruption, developers frequently employ Sedimentation Velocity-Analytical Ultracentrifugation (SV-AUC).
SV-AUC is a first-principles, column-free analytical method that subjects samples to intense centrifugal forces while monitoring sedimentation behavior through UV or interference optics. This technique quantifies monomeric proteins and soluble aggregates directly in their native formulations without introducing matrix interactions. As a result, SV-AUC remains the gold-standard orthogonal method for validating SEC-MALS findings.
For larger particles ranging from 2 μm to 100 μm, Flow Imaging Microscopy has largely replaced traditional Light Obscuration methodologies. Systems such as FlowCam generate high-resolution images of individual particles within a flowing sample stream, allowing simultaneous measurement of concentration, size distribution, and morphology.
Importantly, FIM enables differentiation between genuine protein aggregates and non-protein contaminants, including silicone oil droplets originating from prefilled syringes, degraded polysorbate excipients, and glass particles. Advanced artificial intelligence tools such as VisualAI further enhance analysis by automatically classifying and quantifying subvisible particle populations, supporting batch consistency assessments and reference-product comparability.
Nuanced Conclusions on Aggregation Analysis in Biosimilars
Establishing a robust, multi-attribute analytical platform for aggregation characterization remains one of the most effective strategies for demonstrating biosimilarity while reducing development risk. The integration of advanced mass spectrometry techniques with complementary biophysical methods generates the comprehensive evidence package required by global regulatory authorities.
Today, analytical characterization serves as the primary scientific foundation of successful submissions under both the 351(k) pathway and Article 10(4) regulatory framework. Strong analytical evidence can significantly accelerate development timelines while reducing dependence on large, expensive clinical programs.
Explore our full range of Biosimilar Characterization Services to accelerate your drug development pipeline.
ResolveMass Laboratories Inc. provides advanced mass spectrometry services and comprehensive biophysical characterization solutions designed to meet the stringent expectations of both the FDA and EMA. Through the integration of native mass spectrometry, high-resolution HDX-MS, SEC-MALS, and orthogonal column-free technologies such as SV-AUC, ResolveMass Laboratories Inc. delivers robust comparability data that supports biosimilar development and regulatory success.
Organizations seeking to establish a comprehensive aggregate profiling strategy and strengthen their biosimilar analytical programs can consult the scientific experts at ResolveMass Laboratories Inc. through the Contact Us page.
Frequently Asked Questions about Aggregation Analysis in Biosimilars
Aggregation analysis is regarded as a critical quality attribute because protein aggregates can directly affect the safety, efficacy, and stability of a biotherapeutic product. Even trace levels of aggregates may increase immunogenicity and stimulate the production of anti-drug antibodies (ADAs). These immune responses can reduce therapeutic effectiveness, alter drug behavior in the body, and potentially lead to adverse clinical outcomes. Therefore, aggregate control is an essential component of biosimilar quality assessment.
Native mass spectrometry is designed to maintain proteins in a state that closely resembles their natural solution-phase structure. Instead of using harsh denaturing solvents, the technique employs volatile buffers such as ammonium acetate or ammonium formate under near-physiological conditions. During electrospray ionization, intact protein complexes are transferred gently into the gas phase while preserving important non-covalent interactions. This allows researchers to study native oligomers and aggregates without disrupting their original architecture.
Colloidal aggregates are formed when properly folded protein molecules associate through relatively weak and reversible interactions, such as electrostatic or hydrophobic forces. These assemblies often dissociate when solution conditions are adjusted. Structural aggregates, in contrast, arise when proteins undergo partial unfolding or misfolding, exposing hydrophobic regions that promote strong intermolecular interactions. As a result, structural aggregates are generally more stable, irreversible, and potentially more problematic from a product quality perspective.
Mass spectrometry requires mobile phases that can evaporate cleanly during ionization without leaving behind residues. Traditional phosphate buffers used in conventional SEC contain non-volatile salts that can accumulate within the ion source, causing signal suppression and instrument contamination. Volatile alternatives such as ammonium formate and ammonium acetate are commonly used because they readily vaporize under MS conditions. This compatibility enables direct online coupling between SEC and mass spectrometry while maintaining analytical performance.
SEC-MALS is highly effective for measuring aggregate size distributions and molecular weights, but certain weak or reversible aggregates may dissociate during chromatographic separation. Sedimentation Velocity-Analytical Ultracentrifugation (SV-AUC) addresses this limitation by analyzing proteins directly in their native formulation without column interactions or dilution effects. Because it is a first-principles technique, SV-AUC provides an unbiased assessment of aggregate populations. For this reason, it is frequently used as an orthogonal method to confirm SEC-MALS findings.
HDX-MS provides detailed information about protein conformational dynamics by monitoring hydrogen-deuterium exchange within the protein backbone. Regions that become partially unfolded typically exhibit increased solvent exposure and faster deuterium uptake. As aggregation begins, specific interaction surfaces become protected from the surrounding solvent, resulting in reduced exchange rates. By tracking these localized changes, HDX-MS can identify structural regions that contribute to aggregate nucleation and early self-association events.
Subvisible particles within the 0.1 μm to 10 μm size range are considered particularly important because they can provoke unwanted immune responses. Their size allows them to be efficiently recognized and processed by antigen-presenting cells, potentially stimulating antibody formation. These particles may also present repetitive protein structures that enhance immune system activation. As a result, regulatory agencies require thorough monitoring and characterization of subvisible particle populations throughout product development and stability testing.
The collision cross-section (CCS) value provides information about the three-dimensional shape and size of a protein ion in the gas phase. Unlike mass-to-charge measurements alone, CCS values can distinguish between molecules that have the same mass but different conformations. This additional structural information helps researchers identify compact, folded states as well as extended or partially unfolded conformations. Consequently, CCS measurements are highly valuable for detecting conformational changes associated with aggregation and protein instability.
Reference:
- European Medicines Agency. (2014). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: Quality issues (Revision 1) (EMA/CHMP/BWP/247713/2012). European Medicines Agency. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active-substance-quality-issues-revision-1_en.pdf
- Kaltashov, I. A., Bobst, C. E., Pawlowski, J., & Wang, G. (2020). Mass spectrometry-based methods in characterization of the higher order structure of protein therapeutics. Journal of Pharmaceutical and Biomedical Analysis, 185, 113169. https://doi.org/10.1016/j.jpba.2020.113169
- Cordeiro, M. A., Vitorino, C., Sinogas, C., & Sousa, J. J. (2024). A regulatory perspective on biosimilar medicines. Pharmaceutics, 16(3), 321. https://doi.org/10.3390/pharmaceutics16030321
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (1999). ICH Q6B: Specifications: Test procedures and acceptance criteria for biotechnological/biological products (CPMP/ICH/365/96). European Medicines Agency. https://www.ema.europa.eu/en/ich-q6b-specifications-test-procedures-acceptance-criteria-biotechnological-biological-products-scientific-guideline
- U.S. Food and Drug Administration. (2015, April). Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/135612/download
- Kirchhoff, C. F., Wang, X.-Z. M., Conlon, H. D., Anderson, S., Ryan, A. M., & Bose, A. (2017). Biosimilars: Key regulatory considerations and similarity assessment tools. Biotechnology and Bioengineering, 114(12), 2696–2705. https://doi.org/10.1002/bit.26438

