
Introduction to Data Integrity in Extractables and Leachables Testing
Digital data integrity in extractables and leachables testing refers to the regulatory and scientific requirement that all electronic laboratory records remain complete, accurate, consistent, and reliable throughout their entire lifecycle, from initial generation through final archiving. This requirement ensures that potential chemical migrants originating from packaging materials are accurately detected, characterized, and reported without alteration, manipulation, or loss of critical information. The implementation of robust data integrity practices in extractables and leachables testing has become a central focus for global regulatory authorities committed to protecting patient safety and ensuring product compatibility.
Extractables are chemical compounds that can be forcibly removed from packaging materials, medical devices, or manufacturing systems when exposed to aggressive laboratory extraction conditions. In contrast, leachables are compounds that migrate naturally into a pharmaceutical product under normal storage or use conditions during its shelf life. Accurate characterization of these compounds requires highly sensitive and multidimensional analytical technologies, including Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS), Gas Chromatography-Mass Spectrometry (GC-MS), and Inductively Coupled Plasma Mass Spectrometry (ICP-MS).
To understand the scope of these compounds and their impact, read more about extractables and leachables in pharmaceutical products.
Because these sophisticated analytical platforms rely extensively on computerized data acquisition, peak integration algorithms, and spectral library matching, they generate substantial volumes of dynamic electronic data. Regulatory agencies have repeatedly clarified that considering printed chromatograms as the primary raw data represents a significant misunderstanding of Current Good Manufacturing Practice (cGMP) requirements. Modern compliance expectations require laboratories to secure, validate, preserve, and manage the complete electronic record, including all metadata, processing methods, audit trails, and associated system information.
Share via:
Article Summary:
- Data integrity in E&L testing ensures all electronic records remain accurate, complete, secure, and traceable throughout their lifecycle.
- Advanced analytical tools like LC-MS/MS, GC-MS, and ICP-MS generate large amounts of digital data that must be properly managed and protected.
- Regulations such as FDA 21 CFR Part 11 and EU GMP Annex 11 require validated systems, secure access controls, and audit trails.
- ALCOA+ principles guide compliance by ensuring data is attributable, accurate, original, complete, and readily available.
- Automated peak integration, controlled data processing, and documented changes help prevent data manipulation during chromatographic analysis.
- Regular audit trail reviews are essential to verify data accuracy and detect unauthorized modifications or errors.
- Strong data integrity practices improve patient safety, support regulatory compliance, and reduce the risk of data rejection or approval delays.
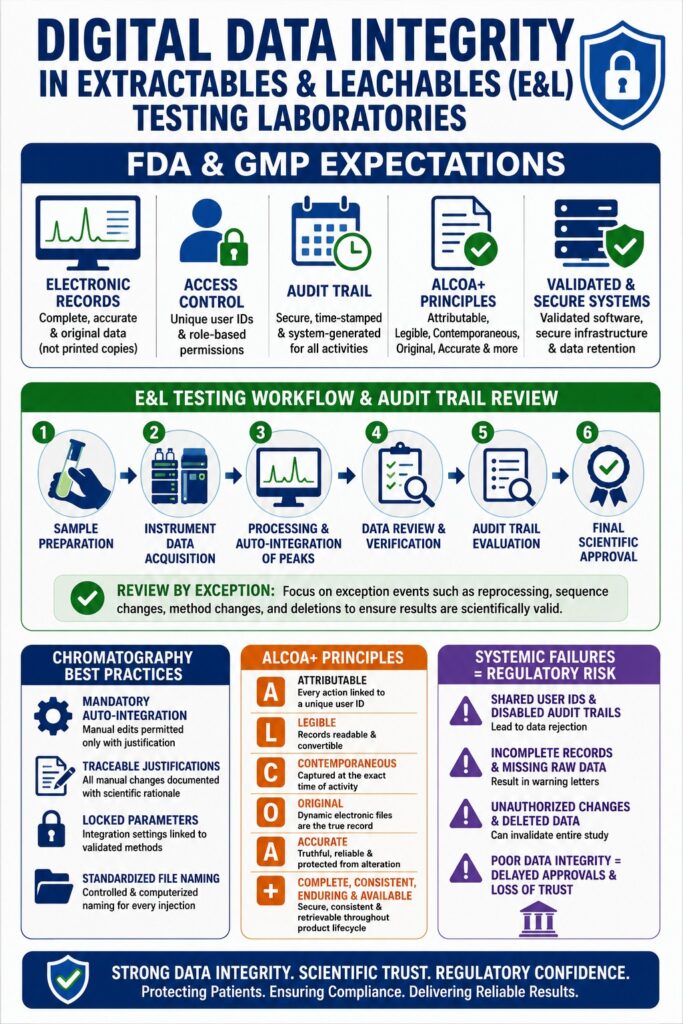
FDA and cGMP Regulatory Expectations for Data Integrity in Extractables and Leachables Testing
Regulatory authorities expect laboratories to validate all computerized systems, implement unique user access controls, and maintain secure, system-generated, time-stamped audit trails. These technical and procedural safeguards ensure that every analytical activity is fully attributable, traceable, and reconstructable during GMP inspections. Regulatory agencies require laboratories to manage data throughout its entire lifecycle, ensuring secure retention and rapid retrieval of raw files and metadata from the point of creation through long-term storage and eventual disposition.
When navigating these requirements, it is essential to consider the usp extractables and leachables standards to ensure full compliance.
To meet international compliance requirements, extractables and leachables testing laboratories must align their computerized systems and workflows with both US FDA 21 CFR Part 11 and EU GMP Annex 11. While FDA 21 CFR Part 11 is a legally binding regulation in the United States that establishes electronic records and electronic signatures as equivalent to paper records, EU GMP Annex 11 serves as the corresponding European guideline emphasizing risk-based control of computerized systems. One notable distinction is that Part 11 specifically requires the capture of initial data creation events within the audit trail, whereas Annex 11 places significant emphasis on documenting changes and deletions involving GMP-relevant records.
Compliance with these frameworks requires the systematic application of the ALCOA+ principles:
Attributable: Every electronic action, from sample injection through peak integration and result generation, must be associated with a unique and verified user identity.
Legible: Electronic records, processing histories, and audit trail information must remain readable and capable of being converted into generally accepted formats throughout the entire retention period.
Contemporaneous: Data must be captured automatically at the exact time analytical activities occur, preventing reconstruction from memory or unauthorized backdating.
Original: The original record must consist of the dynamic electronic file containing all raw spectra, metadata, acquisition parameters, and processing information rather than a static printed representation.
Accurate: Data must be truthful, reliable, error-free, and protected from unauthorized alteration, deletion, or overwriting.
Complete, Consistent, Enduring, and Available: The complete record must contain all analytical activities, including failed, repeated, or aborted runs, maintain consistency across software environments, and remain securely archived and readily retrievable throughout the required product lifecycle.
| Regulatory Framework | Legal Status | Core Electronic Record Scope | Access & Authority Controls | Validation Expectations |
|---|---|---|---|---|
| FDA 21 CFR Part 11 | Binding U.S. Federal Regulation | Requires audit trails documenting record creation, modification, and deletion activities. | Enforces unique user credentials, role-based access control, and account lockout functionality. | Requires comprehensive system validation (IQ/OQ/PQ) demonstrating suitability for intended use. |
| EU GMP Annex 11 | European Union GMP Guideline | Focuses on change management and deletion of GMP-relevant records using a risk-based approach. | Requires physical and logical security controls to identify users and restrict system functions. | Emphasizes qualification of supporting IT infrastructure, networks, and server environments. |
Chromatographic Challenges Influencing Data Integrity in Extractables and Leachables Testing
One of the most significant challenges associated with maintaining data integrity during chromatographic analysis involves the management of complex baselines, peak tailing, and retention time shifts that may encourage subjective manual integration practices. Establishing automatic integration as the mandatory default process, combined with strict procedural oversight, is critical for minimizing these risks. Since extractables and leachables studies frequently involve non-targeted screening of complex polymeric matrices, chromatograms often contain unresolved peaks, elevated baseline noise, and shifting retention profiles that complicate automated quantitation.
To ensure your testing meets the highest standards, find the best cro for extractables and leachables el testing in canada to mitigate these technical risks.
To maintain consistency and analytical reliability, laboratories must establish a comprehensive Standard Operating Procedure (SOP) governing peak integration activities. This SOP should clearly define critical integration parameters, including slope sensitivity, smoothing factors, bunching factors, peak width settings, and reference wavelength requirements. Peak integration represents a fundamental aspect of chromatography because analyte concentration calculations are directly linked to measured peak area. However, because defining peak start and end points can involve subjective judgment, peak integration remains particularly susceptible to manipulation if not properly controlled.
A major compliance concern involves “biased integration,” where analysts apply different integration settings or detection sensitivities to sample analyses compared with calibration standards or system suitability runs. For example, modifying baseline parameters to suppress or exclude small impurity peaks within a stability study may conceal potential leachables that present toxicological risks. To eliminate such vulnerabilities, peak integration procedures must enforce several strict and scientifically justified controls: If you are preparing for specific applications, learn about el testing for inhalation and nasal drug products.
Mandatory Auto-Integration
All chromatographic data must initially be processed using a validated automatic integration algorithm. Manual intervention should be permitted only when validated algorithms cannot adequately resolve co-eluting peaks, severe matrix interference, or complex baseline disturbances.
Traceable Justifications
Every manual peak adjustment must be documented within the electronic audit trail, including a detailed scientific explanation. Justifications should clearly describe the analytical rationale, such as resolving peak tailing caused by an elastomeric stopper extractable, rather than relying on vague or generic descriptions.
Locked Analytical Parameters
Advanced settings such as “integrate inhibit,” “baseline zero,” and “peak masking” must remain linked to validated analytical methods and cannot be modified after data acquisition without formal quality review and approval.
Standardized File Naming
Laboratories must employ a controlled and computerized file-naming convention for every analytical injection, including blanks, system suitability runs, reference standards, and study samples. Informal file names such as “test,” “wash,” or “pre-run” should not be permitted.
Audit Trail Review Workflows for Mass Spectrometry Systems
Audit trail reviews must be conducted systematically by an independent second reviewer to confirm that all modifications, integrations, calculations, and instrument-related events are scientifically justified before final batch release. The incorporation of automated “review by exception” functionality within Chromatography Data Systems significantly reduces review time while maintaining full compliance. The electronic audit trail serves as the definitive chronological record documenting who performed an action, what action occurred, when it occurred, and why it occurred within the analytical system.
Understanding the instrumentation is key; for a deeper look at the technology, explore gc-ms vs lc-ms in extractables and leachables testing.
During an extractables and leachables study, mass spectrometry data is rarely static. A single reported leachable concentration may depend on a complex sequence of interconnected events, including sample injection, instrument tuning, raw ion trace extraction, spectral library matching, and chromatographic peak integration. Reviewers must evaluate the entire sequence to verify that reported results remain scientifically valid and free from manipulation.
Sample Preparation
│
▼
Instrument Data Acquisition
│
▼
Processing and Automatic Integration of Extractable/Leachable Peaks
│
▼
Data Review and Verification
│
▼
Audit Trail Evaluation
│
▼
Final Scientific ApprovalTo simplify this detailed verification process, modern software platforms such as Chromeleon CDS and OpenLab CDS provide advanced filtering, querying, and exception-reporting capabilities. These tools support a highly efficient review-by-exception workflow. When the software confirms that chromatographic peaks were integrated automatically without manual intervention, as indicated by standard baseline codes such as “BB,” reviewers may bypass routine chromatogram inspection and focus their attention on higher-risk activities.
The reviewer’s primary attention should be directed toward exception events, including:
Reprocessing and Recalculation
Reviewing instances in which analytical batches were reprocessed multiple times, identifying parameter changes, and confirming that all recalculations comply with validated SOP requirements.
Sequence and Injection Modifications
Determining whether sample injections, calibration standards, system suitability runs, or blank injections were deleted, reordered, omitted, or excluded from reported results.
Method and Instrument Parameter Changes
Verifying that mass spectrometry acquisition parameters such as gas flows, ionization voltages, collision energies, and detector settings remained unchanged throughout the analytical sequence unless appropriately documented and approved.
Clock Synchronization Integrity
Confirming that system clocks remain synchronized with secure Network Time Protocol (NTP) servers to eliminate the possibility of timestamp manipulation after data acquisition.
Systemic Failures and Regulatory Enforcement Lessons
Regulatory enforcement trends consistently demonstrate that data integrity failures often arise from systemic weaknesses, including shared user credentials, disabled audit trail functionality, and undocumented trial injections. FDA warning letters repeatedly show that these practices can result in complete rejection of analytical data and substantial delays in product approval timelines. Data integrity is not merely a technical compliance requirement; it reflects the overall quality culture, governance structure, and management commitment within a laboratory organization.
Before starting your next study, identify potential pitfalls by reviewing the root causes of failed extractables and leachables el studies.
A review of recent FDA warning letters and Form 483 observations issued to contract testing laboratories, including Shriram Institute, Grace Analytical Laboratory, and Chromatography Institute of America, reveals recurring deficiencies involving computerized systems and record management practices. One of the most frequently cited violations under 21 CFR 211.68(b) involves failure to activate audit trail functionality on chromatography systems or enabling audit trails only immediately before announced inspections. Another common observation under 21 CFR 211.194(a) relates to incomplete laboratory documentation, including discarded raw data sheets, deleted Out-of-Specification (OOS) results, and missing worksheets documenting sample weights and dilution preparations.
| Contract Laboratory & Case | Primary Regulatory Violation Citations | Systemic Data Integrity Failure Mechanism | Regulatory and Commercial Impact |
| Shriram Institute for Industrial Research | 21 CFR 211.68(a) – Failure to exercise appropriate computer system controls | HPLC audit trails remained disabled and were activated only after FDA inspection notification | Formal Warning Letter and mandatory independent third-party audit of laboratory systems |
| Grace Analytical Laboratory Inc. | 21 CFR 211.194(a) – Failure to maintain complete testing records | Analysts recorded data informally before later transcription; missing environmental monitoring logs | Formal Warning Letter and required investigation into informal documentation practices |
| Sanitation & Environment Technology Institute (SDWH) | Failure to provide authentic and valid premarket device safety data | Fabricated testing parameters and copied results from unrelated studies | Immediate rejection of study data, import alerts, and suspension of premarket approvals |
| Mid-Link Technology Testing Co. | Submission of falsified or invalid device testing data | Copied raw data from another study and submitted falsified safety profiles | Rejection of all laboratory-generated data and significant marketing authorization delays |
Scientific Rigor and Technical Excellence in E&L Analytical Campaigns
Developing a compliant extractables and leachables program requires the execution of orthogonal analytical strategies that combine GC-MS, LC-MS/MS, and ICP-MS within a rigorously validated computerized data environment. Applying robust material risk assessment methodologies and utilizing high-resolution analytical technologies ensures that potential contaminants are accurately identified, characterized, and quantified with scientifically defensible precision. A comprehensive E&L investigation depends on the integration of multiple complementary analytical techniques rather than reliance on a single methodology.
Before analytical characterization begins, laboratory scientists conduct a detailed material and component risk assessment based on factors such as route of administration, degree of patient contact, duration of exposure, and material novelty. Products intended for inhalation or injection typically represent the highest clinical risk categories. This assessment guides extraction conditions, solvent selection strategies, and analytical sensitivity requirements.
For those focusing on specific delivery systems, you can also explore specialized testing protocols for el testing for pre-filled syringes.
Material Risk Assessment
│
▼
Selection of Extraction Conditions
│
▼
┌───────────────────────────────────────────────────────────────┐
│ GC-MS │ LC-MS/MS │ ICP-MS │
├────────────────────────┼──────────────────────┼──────────────┤
│ Volatiles & │ Non-volatile │ Elemental │
│ Semi-volatiles │ Organic Compounds │ Impurities │
│ Residual Solvents │ Polar/Non-polar │ Heavy Metals │
│ VOCs │ Organic Residues │ Catalysts │
└───────────────────────────────────────────────────────────────┘
│
▼
Comprehensive Data Review
│
▼
Final Scientific AssessmentTo achieve these demanding chemical characterization objectives, ResolveMass Laboratories Inc. operates a comprehensive, state-of-the-art analytical facility staffed by PhD-level scientists. The laboratory integrates advanced physical and chemical characterization capabilities designed to support complete regulatory compliance and scientific excellence:
High-Resolution Mass Spectrometry (HRMS)
Supports direct infusion and LC-MS analysis for the identification of unknown non-volatile compounds and highly accurate impurity profiling.
Gas Chromatography Platforms (GC-FID and GC-MS)
Utilizes specialized headspace technologies compliant with USP and ICH Q3C requirements for residual solvent analysis and volatile organic compound (VOC) testing.
Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
Provides ultra-trace elemental analysis with sub-parts-per-billion detection capabilities for heavy metals and elemental impurities.
Nuclear Magnetic Resonance (NMR) and Spectroscopy
Enables structural elucidation of complex macromolecules and peptides while supporting Quantitative NMR (qNMR) applications for purity determination.
Thermal and Polymer Characterization
Employs Gel Permeation Chromatography (GPC) for molecular weight distribution analysis and utilizes Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA) for thermal stability and decomposition studies.
Conclusion: The Strategic Value of Data Integrity in Extractables and Leachables Testing
Maintaining data integrity in extractables and leachables testing is both a regulatory requirement and a business-critical necessity that safeguards patient safety, ensures product compatibility, and supports efficient regulatory approval processes. Collaborating with an analytical laboratory that operates a fully validated electronic data management infrastructure ensures that complex chemical characterization studies remain secure, defensible, and ready for regulatory submission. In an environment where global regulators routinely conduct detailed computerized forensic inspections, the scientific credibility of a packaging submission depends entirely on the integrity of the underlying digital data.
By partnering with a contract research organization that incorporates ALCOA+ principles into everyday laboratory operations, pharmaceutical manufacturers significantly reduce the risk of warning letters, data rejection, and regulatory setbacks. This comprehensive compliance framework includes restricted system privileges, synchronized and secured system clocks, automated peak integration protocols, and independent audit trail reviews performed by qualified secondary reviewers before batch approval. Together, these safeguards ensure that every analytical result remains transparent, traceable, scientifically justified, and capable of withstanding the highest levels of regulatory scrutiny.
To discuss an upcoming extractables and leachables study or evaluate compliant analytical workflows for your product’s container closure system, please contact the technical team at ResolveMass Laboratories Inc. through the contact page.
Frequently Asked Questions
Effective computerized system controls are designed to ensure that analytical data remains secure, traceable, and protected from unauthorized changes. Laboratories should implement unique user accounts, role-based permissions, and system-generated audit trails that cannot be altered or disabled. Access to administrative functions must be tightly restricted, and workstation clocks should be synchronized with a secure network time source. Together, these controls help maintain the authenticity and reliability of all electronic laboratory records.
Biased integration occurs when different peak integration settings are applied inconsistently across standards and test samples. Adjustments such as changing baseline placement, peak smoothing, or sensitivity thresholds can significantly alter reported peak areas. As a result, low-level leachables may be overlooked or underreported, creating an inaccurate representation of product safety. Regulatory agencies view such practices as a serious threat to data integrity and scientific validity.
Printed chromatograms and PDF files provide only a static representation of analytical results and do not capture the full electronic record. Critical information such as metadata, instrument settings, processing methods, and audit trail history is typically unavailable in these formats. Regulatory authorities require access to the original electronic files because they allow inspectors to verify data processing activities and reconstruct the complete analytical workflow. Without these records, data transparency cannot be fully demonstrated.
Trial injections, sometimes referred to as pre-injections, are unofficial analytical runs performed before the documented sample sequence begins. These injections are often conducted to evaluate system performance or assess sample results before generating formal records. Regulatory agencies consider this practice problematic because it can create opportunities to conceal unfavorable outcomes, including out-of-specification results. Every analytical activity that contributes to decision-making should be properly documented and included within the official data record.
Review by exception is a risk-based audit trail review approach that focuses attention on unusual or potentially significant system events. Instead of manually reviewing every analytical record, the CDS automatically identifies activities such as manual integrations, data reprocessing, deleted injections, or method modifications. This allows reviewers to concentrate on areas with the greatest compliance risk while maintaining thorough oversight. The approach improves review efficiency without compromising data integrity requirements.
Both regulations emphasize the importance of maintaining secure and traceable electronic records, but their focus differs slightly. FDA 21 CFR Part 11 specifically requires audit trails to document record creation, modification, and deletion activities. EU GMP Annex 11 places greater emphasis on recording changes and deletions that impact GMP-relevant data and often applies a risk-based approach. Understanding these distinctions is important for organizations operating within multiple regulatory jurisdictions.
Chromatographic software used in GxP environments must undergo a structured validation process to demonstrate that it performs reliably and consistently. Laboratories typically develop a Validation Master Plan and User Requirement Specifications before executing Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ). Validation activities should confirm data security, audit trail functionality, backup and recovery capabilities, and processing accuracy. Proper validation ensures the system is suitable for its intended regulatory use.
Accurate time synchronization is essential for preserving the chronological sequence of laboratory activities. Audit trails rely on timestamps to document when analytical actions occur, and these timestamps must accurately reflect real-world events. Synchronizing system clocks with a secure Network Time Protocol (NTP) server helps prevent unauthorized time changes and eliminates the possibility of backdating records. This control strengthens the credibility and traceability of electronic laboratory data.
Reference:
- U.S. Food and Drug Administration. (n.d.). Notifications on data integrity for medical devices. U.S. Department of Health and Human Services. https://www.fda.gov/medical-devices/industry-medical-devices/notifications-data-integrity-medical-devices
- U.S. Food and Drug Administration. (2020, April 15). Shriram Institute for Industrial Research – 597629 – 04/15/2020. U.S. Department of Health and Human Services. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/shriram-institute-industrial-research-597629-04152020
- Blanc, T., Wätzig, H., & Sänger-van de Griend, C. (2025). Peak integration of electropherograms in GMP and research labs: Navigating increased scrutiny amid data integrity audits and inspections. Electrophoresis, 46(11–12), 653–668. https://doi.org/10.1002/elps.70002
- U.S. Food and Drug Administration. (2018, December). Data integrity and compliance with drug CGMP: Questions and answers: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/165535/download
- U.S. Food and Drug Administration. (2025, August 20). Chromatography Institute of America dba Compounder’s International Analytical Laboratory – 708944 – 08/20/2025. U.S. Department of Health and Human Services. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/chromatography-institute-america-dba-compounders-international-analytical-laboratory-708944-08202025
- U.S. Food and Drug Administration. (2019, November 19). Grace Analytical Lab Inc. – 586510 – 11/19/2019. U.S. Department of Health and Human Services. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/grace-analytical-lab-inc-586510-11192019

