
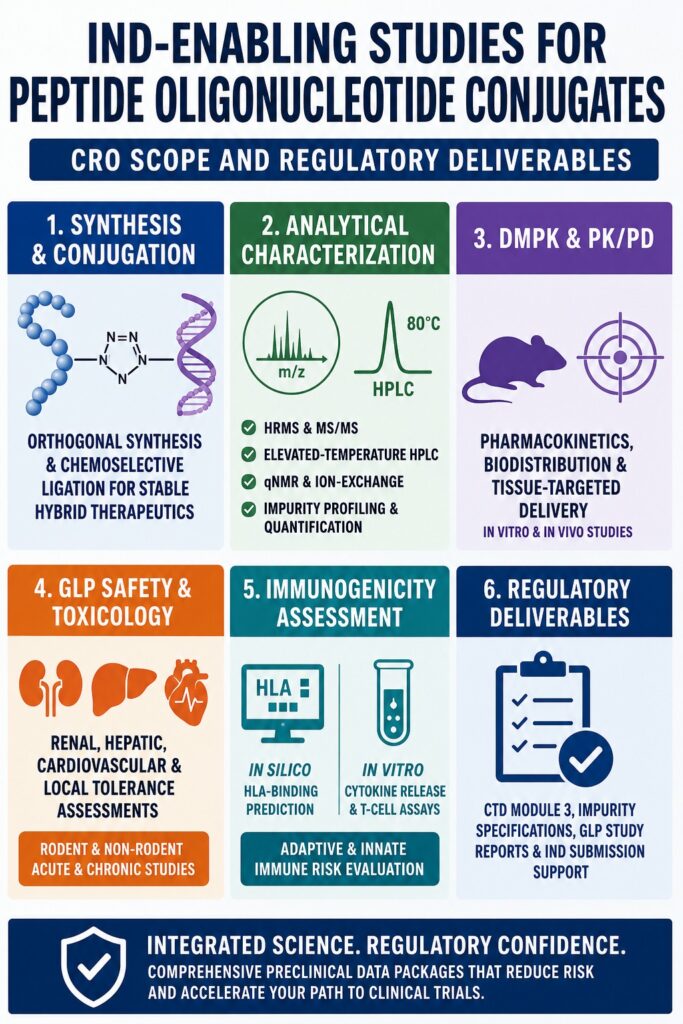
What is the Scope of a CRO in IND-Enabling Studies for Peptide Oligonucleotide Conjugates?
A specialized Contract Research Organization (CRO) plays a critical role in advancing peptide-oligonucleotide conjugates (POCs) from discovery-stage research to clinical development by delivering integrated chemistry, manufacturing, and controls (CMC), pharmacology, and safety assessment services. Advanced CRO infrastructures, including the Montreal-based, ISO 9001:2015-certified and FDA-registered facilities of ResolveMass Laboratories Inc. (Facility ID: 3042696771), support scale-up synthesis, analytical method development and validation, and GLP-compliant toxicology studies necessary to meet international regulatory standards.
Developing a comprehensive preclinical package for hybrid therapeutics requires expertise across multiple scientific disciplines because these compounds combine the targeting precision of synthetic peptides with the gene-modulating properties of oligonucleotides. Successfully integrating these two modalities demands a development partner capable of implementing highly specialized protocols that differ substantially from conventional small-molecule and biologic testing frameworks.
Need expert CMC support for your hybrid therapeutics? Learn more about our integrated CMC services for peptide-oligonucleotide conjugates.
Article Summary:
- CROs support peptide-oligonucleotide conjugate (POC) development through CMC, analytical testing, pharmacology, toxicology, and regulatory services required for IND submissions.
- They develop specialized synthesis and conjugation strategies to overcome chemical compatibility challenges between peptides and oligonucleotides.
- Advanced analytical methods are used to characterize product quality, identify impurities, and ensure manufacturing consistency.
- CROs control process-related contaminants such as residual solvents, heavy metals, and nitrosamines to meet regulatory standards.
- DMPK and PK/PD studies assess biodistribution, tissue targeting, metabolism, and overall therapeutic performance.
- GLP toxicology studies evaluate safety, including kidney, liver, cardiovascular, and local tolerance risks.
- Immunogenicity assessments combine computational and laboratory-based approaches to identify potential immune responses before clinical trials.
How Do CROs Address Synthesis and Chemical Compatibility Challenges?
CROs address the inherent structural and chemical incompatibilities associated with peptide and oligonucleotide synthesis by designing orthogonal protection strategies and employing liquid-phase organic synthesis (LPOS) or parallel automated manufacturing platforms. Synthetic peptides are assembled sequentially from the C-terminus to the N-terminus using Fmoc/tBu chemistry on polystyrene supports, followed by cleavage using strong acidic conditions involving trifluoroacetic acid.
Conversely, synthetic oligonucleotides are synthesized from the 3′ to 5′ direction through phosphoramidite chemistry on controlled pore glass (CPG) supports, requiring basic conditions for final deprotection and cleavage.
To avoid acid-induced depurination of oligonucleotides and base-mediated degradation or racemization of peptides, CROs implement either linear solid-phase assembly (SPOS) approaches or convergent post-synthetic ligation methodologies.
Linear assembly requires carefully engineered protecting group systems, including modified Boc/tBu strategies for side chains such as arginine, aspartic acid, and histidine. These groups can be removed in mild borate buffer conditions at 90°C, thereby avoiding exposure to harsh acidic environments that can damage sensitive nucleic acid backbones.
Convergent synthesis provides a more scalable and efficient alternative. In this strategy, peptide and oligonucleotide fragments are synthesized and purified separately before being chemically linked in a final conjugation step. This approach enables independent optimization of each biomolecular component, decreases synthesis failure rates, and improves process mass intensity (PMI) during manufacturing scale-up.
| Synthesis Parameter | Linear Solid-Phase Assembly | Convergent Post-Synthetic Ligation |
|---|---|---|
| Reaction Phase | Solid support (resin-bound intermediates). | Solution or solid phase (independent fragments). |
| Protecting Group Strategy | Requires specialized, compatible orthogonal groups (e.g., Boc/tBu). | Uses standard, independent protection methods for each fragment. |
| Purification Milestones | Single, complex purification of the final conjugate. | Independent purification of intermediate fragments and final product. |
| Yield and Scaling Limits | Generally limited to shorter sequences because of cumulative cycle losses. | Scalable from milligram quantities to multi-gram clinical production batches. |
| Chemical Stability Risks | Significant risk of depurination or peptide degradation during cleavage. | Minimal risk because assembled hybrids are not exposed to harsh cleavage conditions. |
Explore advanced manufacturing capabilities: Read about our synthesis methods for peptide-oligonucleotide conjugates
and linker chemistry strategies.
What Are the Key Ligation and Conjugation Chemistries for Stable Hybrid Therapeutics?
Chemoselective ligation technologies provide the precise covalent attachment required to create stable hybrid therapeutics while preserving the biological functionality of both the targeting peptide and the gene-silencing oligonucleotide. The chosen bioconjugation method directly influences molecular stability, target-binding affinity, and intracellular release characteristics.
Copper-catalyzed azide-alkyne cycloaddition (CuAAC) is frequently employed to generate highly stable triazole linkers because of its rapid reaction kinetics and excellent yields. However, residual copper must be carefully removed to prevent potential cellular toxicity.
To avoid metal-associated cytotoxicity, strain-promoted azide-alkyne cycloaddition (SPAAC) using dibenzocyclooctyne (DBCO) is commonly selected for therapeutic manufacturing, although it generally proceeds with slower reaction kinetics.
Additional ligation strategies include maleimide-thiol conjugation for forming thioether linkages, oxime ligation, and disulfide bond formation. Disulfide linkers exploit the reducing environment of the intracellular cytosol to enable controlled cleavage, whereas non-cleavable triazole and thioether linkers preserve structural integrity when therapeutic activity depends on maintaining the fully conjugated construct.
What Analytical Deliverables Are Required for CTD Module 3 Impurity Profiling?
Regulatory submissions for CTD Module 3 must include comprehensive characterization of drug substance purity, demonstrating detailed knowledge of impurity profiles, structural integrity, and manufacturing consistency. Because peptide-oligonucleotide conjugates belong to a hybrid therapeutic class, their characterization cannot rely solely on traditional single-modality analytical approaches.
Instead, experienced CROs must establish and validate orthogonal high-resolution analytical methodologies capable of differentiating the desired therapeutic from closely related process-related and product-related impurities. This analytical framework forms the basis of the sponsor’s clinical trial application by supporting scientifically justified specifications and impurity acceptance criteria.
How Are Truncated Sequences and Diastereomers Quantified in IND-Enabling Studies for Peptide Oligonucleotide Conjugates?
CROs identify and quantify sequence-related impurities and stereoisomeric variants through the combined use of high-resolution mass spectrometry (HRMS) and high-performance liquid chromatography (HPLC) performed at elevated temperatures. During automated synthesis, incomplete coupling reactions and side reactions can generate truncated impurities, including n-1 and n-2 deletion sequences, as well as extended n+1 contaminants.
Furthermore, incomplete conjugation can leave residual unconjugated parent species, including free peptide and free oligonucleotide components, which must be accurately resolved and quantified.
The analytical separation of these impurities is often complicated by non-covalent electrostatic interactions between positively charged amino acid residues in the peptide and negatively charged phosphate groups in the oligonucleotide backbone.
To address this challenge, ResolveMass Laboratories Inc. employs elevated-temperature HPLC, typically operated at 80°C, to disrupt secondary structures and electrostatic interactions, producing sharper and more reproducible chromatographic peaks.
Quantification and characterization are performed using a range of validated, highly sensitive analytical technologies:
High-Resolution Mass Spectrometry (HRMS): Instruments employing Orbitrap or Time-of-Flight (TOF) analyzers confirm intact molecular mass with accuracy below 5 ppm, enabling detection of minor chemical modifications.
Tandem MS/MS Sequencing: Collision-induced dissociation (CID) and electron-transfer dissociation (ETD) techniques provide site-specific sequence verification for both peptide and oligonucleotide components.
Quantitative NMR (qNMR): This approach delivers absolute purity measurements for reference standards and active pharmaceutical ingredients without requiring a certified reference standard of the analyte itself, making it particularly valuable for regulatory submissions.
Ion-Exchange Chromatography (IEX): This method separates charge variants and is especially important for characterizing diastereomeric complexity associated with phosphorothioate linkages. For example, the approved antisense oligonucleotide nusinersen contains 17 chiral phosphorothioate centers, producing 2^17 = 131,072 individual diastereomers that require advanced chromatographic and mass spectrometric characterization to demonstrate batch consistency.
| Product-Related Impurity | Origin / Cause | Critical Analytical Detection Method | Regulatory Threshold Strategy |
| Truncated Sequences (n-1, n-2) | Incomplete amino acid or nucleobase coupling cycles. | Elevated-temperature reversed-phase HPLC coupled with HRMS. | Reporting limit typically 0.10%; qualification threshold 1.0%–1.5% based on safety data. |
| Unconjugated Parents | Incomplete conjugation or ester/amide linker cleavage. | Size-exclusion chromatography (SEC) or ion-exchange chromatography (IEX). | Specifications designed to limit free oligonucleotide levels and reduce non-specific accumulation. |
| Diastereomers | Chiral centers in non-natural monomers or phosphorothioate backbones. | High-resolution strong anion-exchange chromatography (AEX). | Characterized as a composite profile and qualified through toxicological batch comparisons. |
| Oxidation and Deamidation | Exposure of peptide residues (e.g., Met, Cys, Asn) to oxygen or extreme pH conditions. | Electrospray ionization mass spectrometry (ESI-MS/MS). | Qualified through forced degradation studies and real-time stability assessments. |
Understand how to optimize your construct: Explore our insights on different types of peptide-oligonucleotide conjugates
and their specific mechanisms of action.
How Are Process-Related Impurities, Nitrosamines, and Heavy Metals Controlled?
Process-related impurities are tightly controlled using gas chromatography and inductively coupled plasma mass spectrometry to ensure compliance with global safety standards. Trace contaminants, including residual solvents such as acetonitrile, dimethylformamide, and pyridine, as well as elemental impurities such as palladium, copper, and nickel catalysts used during ligation reactions, must remain below stringent regulatory limits.
Gas chromatography-mass spectrometry (GC/MS) with headspace sampling remains the industry-standard approach for residual solvent determination and aligns with USP and ICH Q3C recommendations.
Elemental impurity analysis is performed using Inductively Coupled Plasma Mass Spectrometry (ICP-MS) in accordance with USP and ICH Q3D requirements, enabling detection of potentially genotoxic metal contaminants at low parts-per-million (ppm) concentrations.
Additionally, current FDA expectations require a comprehensive nitrosamine risk assessment supported by highly sensitive and validated extraction, detection, and quantification methodologies.
Ensure regulatory compliance: Discover our comprehensive QC testing protocols for peptide-oligonucleotide conjugates.
What DMPK and PK/PD Deliverables Define Preclinical Performance?
Preclinical DMPK and PK/PD studies provide quantitative insights into absorption, tissue distribution, metabolism, and excretion characteristics of peptide-oligonucleotide conjugates. Understanding these biological processes is essential for regulatory submissions because peptide conjugation significantly influences systemic exposure, tissue targeting, metabolic stability, and elimination profiles.
To reduce development risk before first-in-human studies, CROs design extensive in vitro and in vivo ADME programs that support mechanistic pharmacokinetic modeling and translational decision-making.
Optimize your preclinical strategy: Review our specialized preclinical services for peptide-oligonucleotide conjugates.
How Do In Vivo Pharmacokinetic and Biodistribution Studies Support Tissue-Specific Targeting?
In vivo pharmacokinetic and biodistribution investigations demonstrate tissue-selective delivery, cellular internalization, and nuclear localization of oligonucleotide payloads across rodent and non-human primate (NHP) models. Naked oligonucleotides typically undergo rapid clearance and exhibit poor penetration into extrahepatic tissues such as skeletal muscle and the central nervous system (CNS).
By attaching nucleic acids to targeted ligands, including cell-penetrating peptides (CPPs) and receptor-directed peptides, developers can achieve enhanced cellular uptake through receptor-mediated or energy-dependent endocytosis.
Preclinical programs monitor this improved delivery in vivo. In Duchenne muscular dystrophy (DMD) models, peptide-conjugated phosphorodiamidate morpholino oligomers (PMOs) have demonstrated enhanced tissue penetration and uptake.
In non-human primate studies, a single administration of the peptide-PMO conjugate PGN-EDO51 at 30 mg/kg achieved greater than 70% exon 51 skipping in skeletal muscle and diaphragm tissues. This level of activity significantly exceeded that observed with unconjugated PMO formulations and structurally related conjugates.
Similarly, in myotonic dystrophy type 1 (DM1) models, the peptide-conjugate PGN-EDODM1 produced dose-dependent splicing correction in skeletal muscle at doses of 5, 10, and 15 mg/kg, measured using a validated 22-gene expression panel.
To support these studies, CROs validate specialized tissue extraction and quantification procedures, commonly utilizing Solid-Phase Extraction (SPE) and hybridization-ligation ELISA methodologies to measure intact conjugates and active metabolites in tissues including muscle, liver, kidney, heart, and CNS samples.
What Safety Pharmacology and GLP Toxicology Packages Are Required?
GLP safety pharmacology and toxicology packages establish the preclinical safety profile, therapeutic index, and starting dose justification necessary for human clinical studies. These investigations are performed under strict Good Laboratory Practice (GLP) standards and follow internationally recognized guidance documents such as ICH M3, S7A, and S7B.
A complete safety assessment must evaluate both acute and chronic toxicity in one rodent and one non-rodent species, most commonly rats and non-human primates, to support single ascending dose (SAD) and multiple ascending dose (MAD) clinical trial designs.
How Are Renal and Hepatic Liabilities Evaluated in Preclinical Safety Studies?
Preclinical safety programs assess renal proximal tubule accumulation and hepatic enzyme elevations to define safety margins and establish the No Observed Adverse Effect Level (NOAEL). Oligonucleotides and peptide-oligonucleotide conjugates naturally accumulate in the liver and kidneys, making these organs primary sites for potential off-target toxicities.
Within the kidney, oligonucleotides are freely filtered by the glomerulus and subsequently reabsorbed by proximal tubule epithelial cells through megalin-mediated pathways, where they may represent up to 20% of the administered dose. Excessive intracellular accumulation can result in proximal tubule toxicity.
To monitor these risks, CROs evaluate renal function using serum and urinary biomarkers including creatinine, blood urea nitrogen, KIM-1, and NGAL, together with detailed histopathological examination of renal tissue.
Hepatic safety assessments involve monitoring liver transaminases, including alanine aminotransferase and aspartate aminotransferase, as well as evaluating risks associated with hepatic steatosis and liver enzyme elevations that have historically limited some phosphorothioate-based therapies.
In addition, safety pharmacology evaluations must address:
Peptide-Mediated Membrane Perturbation: Cationic cell-penetrating peptides containing high levels of arginine residues, particularly arginine homopolymers such as R11, can disrupt cellular membranes and exhibit substantial toxicity, with LD50 values reported as low as 16.5 mg/kg in mice. Designing constructs with approximately 8–10 arginine residues or incorporating structured cyclic domains can reduce membrane-associated toxicity.
Cardiovascular Safety and QTc Interval Prolongation: Standard GLP safety pharmacology programs require hERG testing and telemetry studies in non-human primates to confirm that the conjugate does not prolong the QTc interval or create proarrhythmic risk.
Local Tolerance and Route-Specific Safety: Toxicology programs must evaluate irritation and inflammatory responses at intended administration sites, including intravenous, subcutaneous, and intrathecal delivery routes.
How Do Evolving Regulatory Guidelines Impact Immunogenicity Assessments?
Alignment with the FDA’s June 2024 final guidance requires implementation of a comprehensive risk-based immunogenicity assessment strategy. This framework evaluates the potential of the conjugate, carrier molecules, and related impurities to induce adverse immune responses.
Because peptide-oligonucleotide conjugates integrate two distinct chemical and biological modalities, they may trigger complex immune responses, including anti-drug antibody (ADA) formation and activation of innate immune pathways.
Sponsors are therefore expected to establish a multi-layered immunogenicity assessment program that begins during candidate selection and continues throughout clinical development.
How Are In Silico and In Vitro Assays Deployed for Immunogenicity Risk Assessment during IND-Enabling Studies for Peptide Oligonucleotide Conjugates?
CROs assess immunogenicity risks through a combination of in silico HLA-binding prediction tools and in vitro cell-based assays designed to evaluate pro-inflammatory cytokine release. Immunogenicity assessment generally consists of two major components: adaptive immune risk evaluation and innate immune activation testing.
In silico platforms evaluate adaptive immune risk by screening peptide carriers, chemical linkers, and junctional regions for potential T-cell epitopes.
These computational approaches estimate binding affinity across a broad range of Human Leukocyte Antigen (HLA) class II alleles, enabling identification of sequences that may promote T-cell activation and subsequent antibody formation.
To validate these computational findings, CROs conduct in vitro studies such as MHC-associated peptide proteomics (MAPPs) and T-cell proliferation assays using human donor PBMCs to determine whether predicted epitopes elicit functional immune responses.
For assessment of innate immune activation, CROs establish cytokine release assays (CRAs). These systems measure drug-induced production of cytokines including IL-6, TNF-α, and IFN-γ in human whole blood or PBMC cultures, providing sensitive detection of sequence-specific or backbone-mediated immune stimulation.
This comprehensive immunogenicity assessment package helps ensure that potentially immunogenic sequence-related impurities are identified, characterized, quantified, and appropriately qualified before the initiation of clinical studies.
Conclusion
The successful completion of IND-Enabling Studies for Peptide Oligonucleotide Conjugates depends on a scientifically rigorous and fully integrated development strategy that combines advanced synthetic chemistry, high-resolution analytical characterization, and comprehensive safety evaluation. Navigating the complex interface between synthetic peptides and therapeutic oligonucleotides requires collaboration with a CRO possessing specialized expertise, robust quality systems, and regulatory-ready analytical capabilities.
Through the implementation of optimized synthesis strategies, elevated-temperature chromatographic methods, and translationally relevant DMPK and GLP toxicology programs, developers can effectively reduce preclinical risks and accelerate regulatory progress.
Ultimately, the generation of a comprehensive and scientifically defensible preclinical data package minimizes development uncertainty and supports a successful transition into clinical evaluation, advancing the clinical development of targeted hybrid therapeutics.
Frequently Asked Questions
During the production of peptide-oligonucleotide conjugates, several degradation mechanisms can affect product quality and stability. Peptides may undergo oxidation, deamidation, amino acid deletion, or racemization, while oligonucleotides can experience depurination, backbone degradation, or hydrolytic cleavage. The contrasting chemical environments required for peptide and oligonucleotide processing often increase the risk of these reactions. As a result, careful process control and monitoring are essential throughout manufacturing.
Peptide-oligonucleotide conjugates often form complex secondary structures due to electrostatic attraction between positively charged peptide residues and negatively charged oligonucleotide backbones. These interactions can lead to poor chromatographic separation, peak broadening, and inaccurate impurity measurements. Running HPLC analyses at elevated temperatures helps disrupt these non-covalent associations and improves peak shape. This approach enables more reliable identification and quantification of closely related impurities.
When synthesizing peptide and oligonucleotide components on the same solid support, CROs must carefully select protecting groups that remain stable throughout multiple reaction conditions. Orthogonal protection strategies are designed so that one group can be removed without affecting another functional group. Modified peptide synthesis schemes are often combined with oligonucleotide-compatible protection chemistry to ensure smooth assembly. This approach minimizes degradation risks and improves overall synthesis efficiency.
Cleavable linkers are specifically engineered to break apart under defined intracellular conditions, releasing the oligonucleotide payload once it reaches the target cell. These linkers may respond to enzymatic activity or reducing environments inside the cell. In contrast, non-cleavable linkers remain intact throughout circulation and tissue distribution, maintaining a permanent connection between the peptide and oligonucleotide. The selection of linker type can significantly influence biodistribution, therapeutic activity, and safety profiles.
Assessment of immunogenicity begins with computational screening tools that identify peptide sequences or linker regions capable of triggering immune responses. These predictions are then supported by laboratory-based studies, including T-cell activation assays and cytokine release testing using human immune cells. Together, these approaches provide insight into both adaptive and innate immune activation. The resulting data help sponsors understand and mitigate potential immunogenicity risks before clinical development.
Phosphorothioate modifications introduce stereochemical complexity by creating chiral phosphorus centers, resulting in large numbers of diastereomeric species. Comprehensive characterization requires advanced analytical technologies capable of separating and identifying these closely related forms. High-resolution ion-pair reversed-phase chromatography, strong anion-exchange chromatography, tandem mass spectrometry, and NMR spectroscopy are commonly employed. Together, these techniques provide a detailed understanding of stereochemical composition and product consistency.
After administration, oligonucleotide-based therapeutics are frequently filtered by the kidneys and subsequently reabsorbed by proximal tubule cells through megalin-mediated pathways. This process can lead to significant intracellular accumulation within renal tissues. Excessive concentrations may contribute to nephrotoxicity and other kidney-related adverse effects. Consequently, preclinical safety studies routinely include biomarker monitoring and histopathological evaluations to assess renal health.
Quantitative NMR (qNMR) serves as a highly accurate analytical tool for determining the absolute purity and concentration of active pharmaceutical materials. Unlike some analytical methods, qNMR does not always require a matching reference standard for the analyte under investigation. This capability makes it particularly valuable for establishing reference materials and verifying analytical accuracy. Regulatory agencies often accept qNMR data as part of chemistry, manufacturing, and controls documentation.
In vitro cytokine release assays are especially important when a therapeutic contains potentially immunostimulatory oligonucleotide sequences or utilizes a novel peptide delivery system. These studies measure the release of inflammatory cytokines from human blood or immune cells following exposure to the drug candidate. The data generated help identify early signs of immune activation and inflammatory risk. Incorporating these assays supports a more comprehensive preclinical safety assessment.
Reference:
- U.S. Food and Drug Administration. (2024, June). Clinical pharmacology considerations for the development of oligonucleotide therapeutics: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-considerations-development-oligonucleotide-therapeutics
- Johannes Winkler (2013). Oligonucleotide conjugates for therapeutic applications. Therapeutic Delivery, 4(7), 791–809. https://doi.org/10.4155/tde.13.47
- Malinowska, A. L., Huynh, H. L., & Bose, S. (2024). Peptide–oligonucleotide conjugation: Chemistry and therapeutic applications. Current Issues in Molecular Biology, 46(10), 11031–11047. https://doi.org/10.3390/cimb46100655
- Gaglione, M., & Messere, A. (2021). Chemistry of peptide-oligonucleotide conjugates: A review. Molecules, 26(18), 5476. https://doi.org/10.3390/molecules26185476
- Pack, B. W. (2024, April 10). Toxicity and immunogenicity considerations for oligonucleotide-related impurities: Impact on control strategy development [Conference presentation]. USP Workshop on Therapeutic Peptides and Oligonucleotides, United States Pharmacopeia. https://www.usp.org/sites/default/files/usp/document/our-work/biologics/documents/brian%20pack_Toxicity%20and%20Immunogenicity%20Considerations.pdf
- Capaldi, D. (2023, February 8). Industry perspective on synthetic oligonucleotide [Conference presentation]. Regulatory and Scientific Virtual Conference on RNA-Based Medicines, European Medicines Agency (EMA). https://www.ema.europa.eu/en/documents/presentation/presentation-industry-perspective-synthetic-oligonucleotide-daniel-capaldi_en.pdf

