
Core Foundations of the Shifting Regulatory Landscape
The regulatory assessment of therapeutic biologics has evolved significantly from a framework centered on independent clinical safety and efficacy studies to a comparative model driven by extensive analytical and functional evidence. This transformation is embedded within abbreviated regulatory pathways, where comprehensive structural characterization determines the extent of clinical investigation required. Unlike small-molecule generics, which are chemically identical versions of their reference products, biologics are highly complex proteins produced through living cellular systems. The importance of this sector is reflected in the rapid growth of the global biosimilar market, which was valued at approximately 26.5 billion in 2024 and is projected by IMARC Group to reach 185.1 billion by 2033, representing a compound annual growth rate (CAGR) of more than 24%.
Ensure your development program meets the highest standards by exploring our comprehensive biosimilar characterization services.
Biological production within clonal cell lines involves a wide range of intricate post-translational modifications (PTMs). These modifications include N-glycosylation patterns at Asn-297 within the Fc region, variations in acidic and basic charge species, the occurrence of high-mannose glycoforms, and terminal clipping of C-terminal lysine residues. Such molecular micro-heterogeneity ensures that different manufacturing processes will inevitably generate detectable structural variations. As a result, regulatory authorities do not require absolute molecular identity. Instead, biosimilars must demonstrate “high similarity with no clinically meaningful differences.” Under this standard, minor differences in inactive components are acceptable provided that they do not affect the product’s potency, purity, safety, or clinical performance.
Learn more about navigating the complexity of post-translational modifications to ensure regulatory compliance.
Article Summary:
- The biosimilar regulatory landscape has shifted from relying primarily on extensive clinical trials to a science-driven model that emphasizes analytical, structural, and functional comparability with a reference biologic.
- Because biologics are produced in living cells and naturally exhibit molecular variability, regulators focus on demonstrating a high degree of similarity rather than requiring exact molecular identity.
- The Totality of Evidence (ToE) framework evaluates biosimilars through multiple layers of evidence, including structural characterization, functional testing, pharmacokinetics/pharmacodynamics (PK/PD), immunogenicity, and, when necessary, confirmatory clinical studies.
- A stepwise development approach reduces uncertainty early through advanced analytical testing, allowing sponsors to streamline non-clinical and clinical programs while maintaining regulatory confidence.
- High-resolution analytical technologies, particularly mass spectrometry and complementary biophysical methods, play a critical role in identifying molecular attributes, post-translational modifications, and quality characteristics that determine biosimilarity.
- Functional assays and receptor-binding studies verify that the biosimilar performs the same biological activities as the reference product, ensuring comparable efficacy, safety, and mechanism of action.
- Recent regulatory updates from agencies such as the FDA and EMA increasingly support reduced clinical trial requirements when robust analytical and PK/PD evidence is available, lowering development costs, accelerating approvals, and reinforcing the importance of comprehensive analytical characterization in biosimilar submissions.
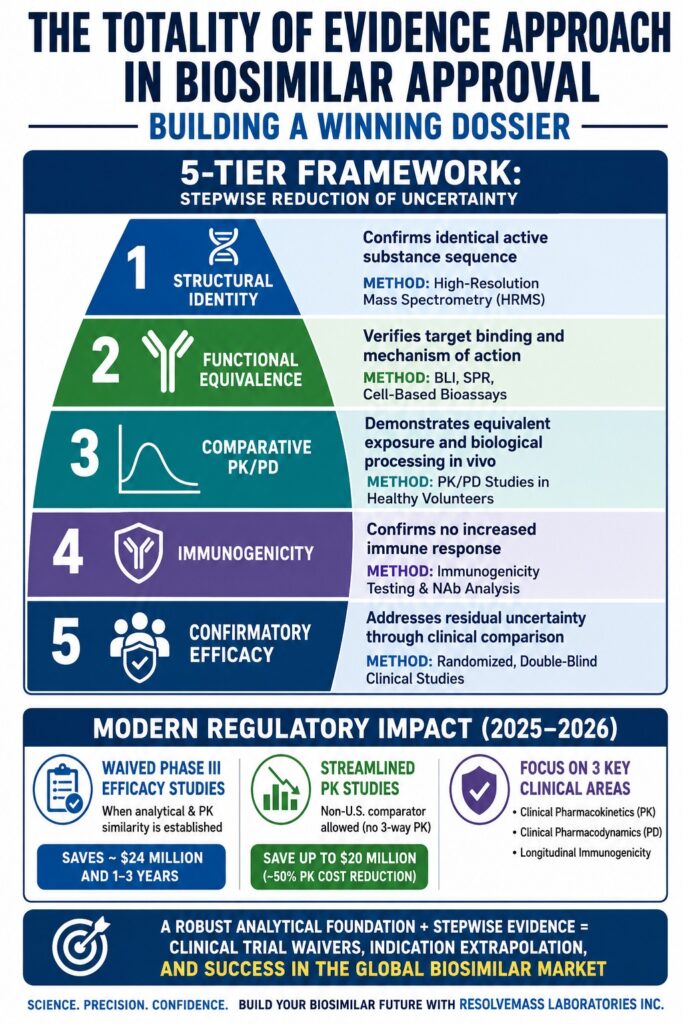
Structural and Functional Alignment in the Totality of Evidence Approach in Biosimilar Approval
The Totality of Evidence Approach in Biosimilar Approval is a comprehensive, multidisciplinary framework that evaluates a candidate biosimilar through the combined assessment of structural, functional, pharmacokinetic, and immunogenic characteristics relative to a single reference product. Within this framework, regulatory approval is not dependent on the outcome of any individual study. Instead, approval is based on the cumulative evidence generated through a stepwise reduction of uncertainty across analytical and clinical domains. By establishing a high degree of similarity in both structural and biological properties, sponsors create a scientific bridge that enables reliance on the existing safety and efficacy data of the originator biologic.
| Pyramid Layer | Target Parameter | Primary Analytical/Clinical Methodology | Regulatory Purpose & Impact |
|---|---|---|---|
| Layer 1: Structural Identity | Amino acid sequence, intact mass, and primary structure | High-Resolution Mass Spectrometry (HRMS) | Confirms identical active substance sequence relative to the reference product |
| Layer 2: Functional Equivalence | Receptor binding kinetics, signaling activation, and cell-based potency | Biolayer Interferometry (BLI), Surface Plasmon Resonance (SPR), and cell-based bioassays | Verifies target binding and mechanism of action |
| Layer 3: Comparative PK/PD | Systemic exposure, half-life, clearance, and biodistribution | Comparative pharmacokinetic and pharmacodynamic studies in healthy volunteers | Demonstrates equivalent biological processing and exposure in vivo |
| Layer 4: Immunogenicity | Anti-drug antibodies (ADAs) and neutralizing antibodies (NAbs) | Longitudinal immunogenicity testing and neutralizing antibody characterization | Confirms no increased immune response compared with the reference product |
| Layer 5: Confirmatory Efficacy | Clinical safety, objective response rates (ORR), and progression-free survival (PFS) | Randomized, double-blind comparative clinical studies in sensitive patient populations | Addresses residual uncertainty not resolved through analytical assessment |
Discover how our team defines and verifies critical quality attributes for your drug product.
Stepwise Data Generation Under the Totality of Evidence Approach in Biosimilar Approval
Stepwise data generation provides a structured, risk-based development pathway in which findings from analytical characterization directly influence the design and scope of subsequent clinical investigations. By systematically reducing uncertainty at highly sensitive analytical stages, sponsors can justify abbreviated non-clinical and clinical development programs.
Build a robust submission package by leveraging our expert biosimilar comparability studies.
When structural analyses identify a minor discrepancy in a post-translational modification, developers must specifically investigate that variation using functional bioassays or pharmacokinetic studies to demonstrate the absence of biological consequences. For instance, the regulatory approval pathways of the infliximab biosimilar PF-06438179/GP1111 (Ixifi/Zessly) and the Sandoz pegfilgrastim biosimilar (LA-EP2006, Ziextenzo) successfully relied on this stepwise confirmation model. These products obtained approval across all eligible indications in both the United States and Europe without requiring separate clinical trials for every therapeutic indication.
Analytical Characterization: Deconstructing the Molecular Fingerprint
Physicochemical and structural characterization represents the most sensitive component of a biosimilar application, capable of identifying micro-heterogeneities that conventional clinical trials are statistically unable to detect. Developing a robust comparative analytical assessment (CAA) requires multiple orthogonal analytical techniques to demonstrate that the biosimilar’s molecular profile falls within the established lot-to-lot variability of the originator product.
Operating from a state-of-the-art facility in Montreal, Canada, while supporting pharmaceutical organizations throughout the United States, Europe, and Asia, ResolveMass Laboratories Inc. provides extensive analytical expertise through advanced high-resolution mass spectrometry and polymer characterization services. The laboratory functions as an FDA-registered testing facility (FDA Establishment Identifier No. 3042696771) and maintains ISO 9001:2015 certification. Its analytical workflow incorporates a multi-tier validation strategy for every dataset, including system suitability testing using human monoclonal antibody reference materials such as SigmaMAb™ to ensure full traceability and data integrity.
| Quality Domain | Specific Target Parameter | Analytical Platform & Method |
| Primary Structure | Verification of peptide sequence identity and disulfide bond configurations | High-resolution LC-MS/MS peptide mapping and intact mass deconvolution |
| Glycan Profile | Quantification of N-glycosylation, core fucosylation, and terminal sialylation | HILIC-UPLC with Fluorescence Detection (FLD) and online mass spectrometry |
| Higher-Order Structure | Characterization of secondary, tertiary, and quaternary conformations | Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) and Quantitative NMR |
| Heterogeneity & Size | Identification of aggregates, fragments, and molecular weight variants | Gel Permeation Chromatography (GPC), SEC-MALS, and Multi-Angle Light Scattering |
| Thermal Properties | Assessment of glass transition temperatures, melting behavior, and structural stability | Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA) |
Gain precise insights into your product’s structure with our advanced peptide mapping.
High-Resolution Mass Spectrometry (HRMS) Capabilities
At the center of the bioanalytical characterization package is the application of advanced Orbitrap and Time-of-Flight (Q-TOF) mass spectrometry technologies. Conventional mass spectrometry platforms often lack the resolving power necessary for precise characterization of intact therapeutic proteins. High-resolution mass spectrometry provides sub-Dalton mass accuracy with mass error ranges typically between ±1 and 5 parts per million (ppm), enabling developers to confirm molecular formulas, map peptide sequences, and quantify low-abundance glycoforms with exceptional precision.
This analytical capability is essential when comparing highly complex biomolecules such as trastuzumab, bevacizumab, and ustekinumab against their corresponding reference products. The technology allows for the detection and characterization of even subtle post-translational modifications that could influence biosimilarity assessments.
Discover how native mass spectrometry provides superior structural insights without disrupting non-covalent interactions.
Advanced Biophysical Verification: Binding Kinetics and Functional Assays
Advanced biophysical characterization and cell-based bioassays are essential for confirming that a biosimilar exhibits physiological binding activity consistent with the reference product’s established mechanism of action. These studies verify that the structural integrity of the molecule translates into equivalent biological potency and expected clinical performance. For monoclonal antibody (mAb) biosimilars, comprehensive functional testing evaluates both the variable Fab domain responsible for antigen binding and the Fc domain responsible for effector functions and pharmacokinetic regulation.
Biolayer interferometry (BLI) panels have become particularly important for evaluating Fc-mediated effector functions. A complete BLI assessment for a monoclonal antibody biosimilar includes seven critical binding parameters: antigen interaction kinetics, neonatal Fc receptor (FcRn) binding, the complete activating and inhibitory Fc-gamma receptor (FcγR) profile, and complement activation through C1q binding.
To reduce analytical artifacts and maintain data quality, loading density must be carefully controlled. For example, when using a His XT biosensor, CD32a (FcγRIIa) loading should remain below a 0.5 nm shift to prevent aggregate-related interference and distortion of kinetic measurements. In addition, during C1q binding studies, immobilization of the antibody through its light chain ensures that the Fc domain remains fully exposed and accessible. This strategy avoids amine coupling approaches that may alter molecular conformation and produce misleading negative binding results.
Beyond receptor-binding assessments, cell-based functional assays examine downstream signaling pathways, including antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). When these cellular mechanisms constitute the primary mode of therapeutic activity, demonstrating functional equivalence in vitro becomes a critical component of the biosimilar development program. These studies connect structural characterization with in vivo pharmacokinetic outcomes, confirming that minor post-translational variations do not adversely affect clinical efficacy or safety.
Ensure comprehensive quality control by utilizing our charge variant analysis.
Modernizing Clinical Strategies: Wave of Waived Comparative Efficacy Studies
Recent updates from global regulatory authorities have substantially modernized biosimilar development by permitting the waiver of traditional Phase III comparative efficacy studies when sufficient analytical and pharmacokinetic similarity has been demonstrated. These regulatory advancements, introduced during late 2025 and early 2026, have significantly reduced development costs while accelerating market entry.
In draft guidance released in October 2025 and further expanded through revised Q&A documents in March 2026, CDER Commissioner Marty Makary, M.D., M.P.H. announced a major initiative aimed at simplifying biosimilar licensing through the reduction of unnecessary clinical studies. The agency emphasized that comparative efficacy studies (CES), which frequently require one to three years to complete and cost approximately 24 million, are often less sensitive than modern analytical technologies when identifying subtle molecular differences.
The revised guidance also permits sponsors to streamline clinical pharmacokinetic (PK) studies by using a non-U.S.-licensed comparator product without conducting a three-way PK bridging study. This change can reduce pharmacokinetic trial expenses by as much as 50%, resulting in potential savings of approximately 20 million, provided sufficient analytical and functional bridging data are available.
This regulatory evolution not only lowers development costs but also addresses ethical concerns associated with conducting large-scale, redundant clinical trials in human subjects. Under this streamlined development model, clinical programs concentrate on three key areas:
Clinical Pharmacokinetics (PK): Assessment of systemic exposure and absorption characteristics in healthy volunteers to establish bioequivalence.
Clinical Pharmacodynamics (PD): Measurement of relevant biomarkers that reflect the therapeutic mechanism and downstream biological response.
Longitudinal Immunogenicity: Evaluation of anti-drug antibody (ADA) formation to ensure that the biosimilar does not trigger an increased immune response relative to the reference product.
The practicality of this approach has been demonstrated by Sandoz pegfilgrastim, which achieved close alignment with its reference product across structural and functional evaluations, resulting in pharmacokinetic and pharmacodynamic similarity in healthy volunteers and comparable safety outcomes in sensitive oncology populations. Likewise, the bevacizumab biosimilar MB02 demonstrated equivalent efficacy, achieving an objective response rate risk ratio of 0.910 and a comparable safety profile in advanced non-small cell lung cancer (NSCLC) during the STELLA study. These findings further support the predictive value of robust analytical characterization in establishing clinical equivalence.
Impurity Profiling and Lifecycle Control in the CMC Dossier
The management of manufacturing-related impurities and lifecycle process modifications remains a fundamental regulatory requirement for ensuring that structural changes do not introduce unexpected immunogenicity risks. A compliant Chemistry, Manufacturing, and Controls (CMC) dossier must incorporate highly sensitive analytical methodologies capable of quantifying process-related and product-related impurities at parts-per-billion or parts-per-trillion concentrations.
Process-related impurities present a significant safety concern, particularly when alternative host cell expression systems are used during biosimilar production. These impurities include host cell proteins, residual host cell DNA, residual solvents, carcinogenic nitrosamines, and extractables and leachables (E&L) originating from single-use bioreactor systems and container-closure components. Environmental contaminants, including per- and polyfluoroalkyl substances (PFAS), must also be monitored carefully to ensure compliance with regulatory expectations.
Product-related impurities, particularly aggregate species, require strict control through advanced analytical techniques such as size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) to minimize the potential for immunogenic reactions.
Under the FDA’s proposed three-tier framework, critical quality attributes (CQAs) are classified according to their potential impact on clinical outcomes. Tier 1 attributes, which represent the highest level of clinical risk, are subjected to rigorous statistical equivalence testing. These analyses utilize multiple reference product lots to establish acceptable variability ranges. As discussed by Chow et al. and Wang and Chow, evaluation of multiple test samples per lot can generate an unbiased estimate of reference variance (σR) under conventional single-sample methodologies. However, when multiple samples are collected from individual lots, specialized statistical adjustments are required to prevent bias and appropriately account for inter-lot heterogeneity.
Maintain the highest safety standards with our specialized impurity profiling.
[ Raw Material Input ]
│
▼
[ Clonal Cell Culture Expansion ]
- Parameter Controls: pH, O₂, Temp
│
▼
[ Purification Operations ]
- Column Chromatography (Protein A, IEX)
│
┌──────────────────────────┴──────────────────────────┐
▼ ▼
[ Product Quality Monitoring ] [ Impurity Profile Assessment ]
• Glycosylation Profiling (Asn-297) • Host Cell Proteins (HCPs) & DNA
• Charge Heterogeneity (cIEF) • Nitrosamines & PFAS Analysis
• Higher-Order Conformation (HDX-MS) • Extractables & Leachables (E&L)
│ │
└──────────────────────────┬──────────────────────────┘
▼
[ Final Drug Product Lot ]
- Validation of multi-lot distribution limitsAchieving this level of analytical rigor requires access to highly specialized testing capabilities. ResolveMass Laboratories Inc. addresses these demands through a comprehensive suite of bioanalytical services, including Extractables and Leachables (E&L) testing, high-resolution impurity profiling, and ultra-sensitive detection of Nitrosamines and PFAS across diverse sample matrices. Operating from a US FDA-registered and ISO 9001:2015-certified facility, ResolveMass delivers the scientific traceability, multi-tier data validation, and analytical confidence necessary to support global CMC compliance and defensible regulatory submissions.
Conclusion
Successful implementation of the Totality of Evidence Approach in Biosimilar Approval depends on building an analytically driven dossier that effectively connects structural characterization with biological function. By establishing a high degree of similarity and leveraging advanced biophysical assessment tools, developers can confidently pursue clinical trial waivers, support indication extrapolation strategies, and strengthen their position within the global biosimilar marketplace.
Emerging regulatory frameworks, including the 2026 FDA and EMA draft guidance updates, increasingly reward organizations that prioritize highly sensitive analytical characterization rather than relying heavily on extensive clinical efficacy studies. This shift creates opportunities for development savings of up to 44 million per biosimilar asset while maintaining rigorous scientific standards.
The foundation of a successful biosimilar submission remains a comprehensive analytical dataset capable of mapping the molecular fingerprint of the biosimilar against the established lot-to-lot variability of the reference biologic. Achieving this level of scientific precision is greatly enhanced through collaboration with contract research organizations that possess advanced Orbitrap HRMS technologies and validated analytical methodologies. ResolveMass Laboratories Inc. offers the regulatory expertise, high-resolution mass spectrometry capabilities, and multi-tier data validation systems necessary to support the development of a compliant BLA dossier.
To optimize your biosimilar characterization strategy and discuss analytical comparability requirements, connect directly with the scientific team through the ResolveMass Contact Page.
Frequently Asked Questions
The comparative analytical assessment is designed to establish that a biosimilar closely matches its reference biologic in terms of structural, physicochemical, and biological characteristics. Advanced analytical technologies are used to compare critical quality attributes and identify any potential differences. This evidence forms the foundation of the biosimilar development program and helps regulators determine whether additional clinical testing can be minimized. A strong analytical package significantly reduces uncertainty regarding product similarity.
The 351(a) pathway is intended for original biologic products and requires comprehensive clinical and non-clinical evidence to independently demonstrate safety, efficacy, and quality. In contrast, the 351(k) pathway is specifically designed for biosimilars and relies on demonstrating a high degree of similarity to an already approved reference product. Rather than repeating extensive clinical development, sponsors use comparative analytical, functional, and pharmacokinetic data to support approval. This approach streamlines development while maintaining regulatory standards.
Yes, current regulatory guidance allows the use of certain non-U.S.-licensed comparator products during biosimilar development programs. This flexibility can simplify global development strategies and reduce the need for duplicative studies. However, developers must establish a robust analytical and functional bridge between the non-U.S. comparator and the U.S.-licensed reference product. When sufficient scientific evidence supports the relationship between the products, additional bridging studies may not be necessary.
A comprehensive analytical panel for monoclonal antibody biosimilars should evaluate several critical binding interactions associated with the product’s mechanism of action. These assessments typically include antigen-binding affinity, neonatal Fc receptor (FcRn) interactions, activating and inhibitory Fc-gamma receptor (FcγR) binding, and complement protein C1q engagement. Together, these measurements provide a detailed understanding of both Fab-mediated and Fc-mediated functions. Evaluating these parameters helps confirm functional similarity with the reference product.
Comparative clinical efficacy studies may be waived when extensive analytical and functional evidence demonstrates that the biosimilar is highly similar to the reference product. Regulatory authorities may also consider comparable pharmacokinetic and pharmacodynamic data when evaluating waiver requests. The goal is to avoid unnecessary clinical trials when sensitive analytical methods have already addressed potential uncertainties. This modern approach helps reduce development costs and accelerates patient access to biosimilar therapies.
High-Resolution Mass Spectrometry (HRMS) plays a central role in evaluating biosimilarity because it provides highly accurate molecular characterization of therapeutic proteins. The technology is used to verify amino acid sequences, identify post-translational modifications, and assess molecular heterogeneity. Advanced platforms such as Orbitrap and Q-TOF instruments can detect even subtle structural differences with exceptional precision. These capabilities make HRMS one of the most powerful analytical tools in biosimilar development.
Scientific justification enables regulators to extend approval of a biosimilar beyond the indication directly studied in clinical trials. To support extrapolation, developers must demonstrate that the biosimilar and reference product share the same mechanism of action, pharmacokinetic behavior, biodistribution profile, and safety characteristics across all proposed indications. Comprehensive analytical and functional evidence is essential for this process. When supported by robust data, extrapolation can eliminate the need for multiple indication-specific clinical studies.
An interchangeable biosimilar is a product that meets additional regulatory requirements beyond those required for standard biosimilar approval. Manufacturers must demonstrate that the biosimilar can be expected to produce the same clinical outcome as the reference biologic in any patient. In many cases, switching studies are conducted to evaluate the impact of alternating between the biosimilar and the reference product. Once designated as interchangeable, the product may be substituted at the pharmacy level according to applicable regulations.
Reference:
- U.S. Food and Drug Administration. (2025). Considerations in demonstrating biosimilarity to a reference product: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/182186/download
- U.S. Food and Drug Administration. (2026, March 9). FDA takes further steps to streamline biosimilar development and make medicines more affordable. https://www.fda.gov/news-events/press-announcements/fda-takes-further-steps-streamline-biosimilar-development-and-make-medicines-more-affordable
- Derzi, M., Johnson, T. R., Shoieb, A. M., et al. (2019). The “totality-of-the-evidence” approach in the development of PF-06438179/GP1111, an infliximab biosimilar, and in support of its use in all indications of the reference product. Advances in Therapy, 36(4), 805–823. https://doi.org/10.1007/s12325-019-0884-4
- Hung, A., Vu, Q., & Mostovoy, L. (2017). A systematic review of U.S. biosimilar approvals: What evidence does the FDA require and how are manufacturers responding? Journal of Managed Care & Specialty Pharmacy, 23(12), 1234–1244. https://doi.org/10.18553/jmcp.2017.23.12.1234
- Markus, R., Liu, J., Ramchandani, M., Landa, D., Born, T., & Kaur, P. (2017). Developing the totality of evidence for biosimilars: Regulatory considerations and building confidence for the healthcare community. BioDrugs, 31(3), 175–187. https://doi.org/10.1007/s40259-017-0218-5
- European Medicines Agency. (2014). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: Quality issues (Revision 1) (EMA/CHMP/BWP/247713/2012). European Medicines Agency. https://www.ema.europa.eu/en/similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active-substance-quality-issues-scientific-guideline
- Liu, J., Eris, T., Li, C., Cao, S., & Kuhns, S. (2017). Assessing analytical similarity of proposed biosimilar products: A review of statistical approaches. Clinical Pharmacology & Therapeutics, 101(6), 749–759. https://doi.org/10.1002/cpt.651

