
Introduction
Protein Binding and Free Fraction Measurement in Bioanalysis is a fundamental analytical process used to determine the concentration of unbound drug that is available to cross biological membranes, interact with pharmacological targets, and undergo metabolic or renal elimination. Measuring this unbound concentration establishes the physiological connection between in vitro pharmacological activity and in vivo clinical performance. As a result, it plays a direct role in determining appropriate clinical dosing strategies, defining toxicological safety margins, and evaluating the risk of drug-drug interactions (DDIs). According to the free drug hypothesis, only the unbound (free) fraction (f_u) of a drug circulating in the systemic bloodstream can distribute into tissues and produce either therapeutic or toxicological effects. The concentration of this pharmacologically active fraction is controlled by the reversible binding of drugs to major plasma proteins, primarily human serum albumin (HSA) for acidic and neutral compounds, and α₁-acid glycoprotein (AGP) for basic compounds. Therefore, implementing a scientifically rigorous approach to Protein Binding and Free Fraction Measurement in Bioanalysis is essential for accurately characterizing complex absorption, distribution, metabolism, and excretion (ADME) properties throughout the drug discovery and development process.
Learn how to build a winning bioanalytical strategy for your drug development program to ensure regulatory success.
In the past, early-stage drug candidates were frequently assessed using conservative assumptions. One common practice involved assigning a default reportable free fraction of 0.01 (1%) whenever plasma protein binding exceeded 99%. However, advances in modern drug discovery have led to the development of highly lipophilic small molecules, covalent inhibitors, peptides, and antisense oligonucleotides (ASOs) that often exhibit exceptionally strong plasma protein binding, producing true free fractions that are substantially lower than 1% (f_u < 0.01). Accurately measuring these extremely small unbound fractions demands bioanalytical methodologies with outstanding analytical sensitivity, strict control of temperature and pH conditions, and carefully matched biological matrices to minimize systematic analytical bias. As a leading provider of high-precision bioanalytical solutions, ResolveMass Laboratories Inc. addresses these analytical challenges by combining advanced high-sensitivity liquid chromatography-tandem mass spectrometry (LC-MS/MS) with carefully optimized separation methodologies. Through rigorous control of thermodynamic equilibrium conditions and comprehensive recovery assessments, ResolveMass Laboratories Inc. generates validated, submission-ready datasets that comply with the increasingly stringent expectations of global regulatory authorities.
Discover the critical steps required to consistently generate robust bioanalytical data for your most complex assays.
Share via:
Article Summary:
- Protein binding determines pharmacological activity: Only the unbound (free) fraction of a drug is capable of reaching target tissues, producing therapeutic effects, undergoing metabolism, and being eliminated. Therefore, accurate free fraction (f_u) measurement is essential for reliable pharmacokinetic and pharmacodynamic assessments.
- Multiple analytical techniques are available: Equilibrium dialysis remains the regulatory gold standard, while ultrafiltration, ultracentrifugation, flux dialysis, and solid-phase microextraction (SPME) offer alternative approaches. The most suitable method depends on the drug’s physicochemical properties, stability, protein-binding characteristics, and analytical objectives.
- Experimental conditions strongly influence accuracy: Factors such as osmotic volume changes, non-specific adsorption to laboratory materials, membrane interactions, temperature, and plasma pH can introduce significant bias. Careful control of these variables is necessary to obtain reproducible and scientifically reliable results.
- Modern analytical technologies improve low-level detection: Highly sensitive LC-MS/MS platforms, optimized sample preparation, matrix-matched calibration, and improved separation techniques enable accurate quantification of drugs with extremely low free fractions, including compounds with protein binding greater than 99.9%.
- Regulatory guidance now supports experimentally measured free fractions: Current ICH M12 recommendations allow the use of validated free fraction values below 1% instead of relying on historical default assumptions, leading to more realistic drug-drug interaction (DDI) predictions and better-informed regulatory decisions.
- Comprehensive bioanalytical validation is essential: Protein-binding assays must demonstrate acceptable accuracy, precision, selectivity, calibration performance, and control of matrix effects in accordance with ICH M10 principles to ensure data integrity and regulatory acceptance.
- Validation should match the intended application: Fit-for-purpose qualification may be sufficient during early drug discovery, whereas GxP-compliant validated methods are expected for IND-enabling studies and clinical development. Specialized analytical strategies are also required for complex therapeutic modalities such as antisense oligonucleotides, siRNA therapeutics, and monoclonal antibodies.
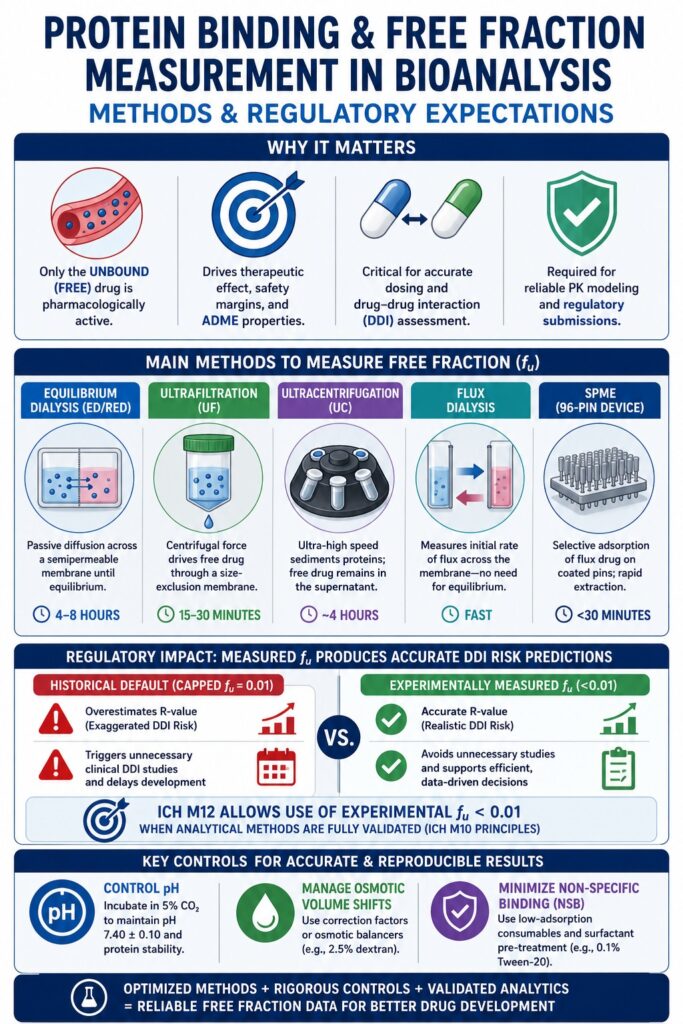
What are the Main Experimental Methods for Protein Binding and Free Fraction Measurement in Bioanalysis?
Several analytical techniques are routinely employed to separate and quantify the unbound fraction of drugs present in biological matrices. The most widely used approaches include equilibrium dialysis, ultrafiltration, ultracentrifugation, flux dialysis, and solid-phase microextraction (SPME). These methodologies isolate free drug molecules from protein-bound complexes through mechanisms such as physical size-exclusion membranes, ultra-high-speed centrifugal sedimentation, or selective adsorption onto specialized extraction surfaces. Choosing the most appropriate analytical platform depends on multiple factors, including the physicochemical characteristics of the compound, its stability within the biological matrix, the anticipated free-fraction range, and its tendency to undergo non-specific binding.
Explore the benefits of establishing a dedicated bioanalytical CRO partnership to streamline your analytical studies.
Equilibrium Dialysis (Thermodynamic Standard)
[Plasma + Drug] | [Phosphate-Buffered Saline]
|
| <--- Semi-Permeable Membrane --->
| (Passive diffusion of free
| drug until equilibrium)
Equilibrium Dialysis and Rapid Equilibrium Dialysis (RED)
Equilibrium dialysis determines the unbound fraction of a drug by permitting free drug molecules to diffuse passively through a semipermeable membrane that separates a plasma-containing donor chamber from a buffer-containing receiver chamber. Modern Rapid Equilibrium Dialysis (RED) systems have significantly improved this traditional technique by incorporating automated 96-well plate formats, reducing the required incubation period to approximately 4 to 6 hours. The semipermeable membrane is designed with a defined molecular weight cut-off (MWCO), generally ranging from 8 to 12 kDa, allowing small drug molecules to pass freely while retaining large plasma proteins such as albumin (~66 kDa) and AGP (~40 kDa) within the donor compartment. Diffusion continues until thermodynamic equilibrium is reached, at which point the concentration of free, unbound drug becomes identical on both sides of the membrane. The fraction unbound (f_u) is then calculated using the following thermodynamic relationship:
fu = Cbuffer / Cplasma
Where:
- Cbuffer represents the drug concentration measured in the buffer compartment.
- Cplasma represents the total drug concentration measured in the plasma compartment.
Understand the critical role of stability testing in bioanalysis to prevent sample degradation and ensure accurate quantification.
Ultrafiltration and Methodological Divergence
Ultrafiltration separates the free drug fraction by applying centrifugal force to drive the aqueous phase of plasma through a size-exclusion membrane, creating a rapid, non-equilibrium separation process. This method is particularly advantageous when analyzing chemically unstable compounds because it minimizes sample processing time. However, ultrafiltration remains susceptible to several methodological limitations, including temperature-dependent variability, membrane fouling, and systematic analytical bias.
During the procedure, plasma containing the drug is transferred into a filtration device equipped with a membrane possessing a defined MWCO, typically between 10 and 30 kDa. Low-speed centrifugation forces the aqueous ultrafiltrate containing unbound drug molecules through the membrane, while protein-bound drug complexes remain above the filter.
To minimize concentration polarization, a phenomenon in which plasma proteins accumulate near the membrane surface and disrupt the equilibrium between bound and unbound drug, the volume of ultrafiltrate collected should remain below 10% of the original plasma sample volume. Although ultrafiltration offers substantial advantages in speed, it may also introduce systematic analytical errors. For example, measurements of the unbound fraction of ceftriaxone obtained using ultrafiltration have been shown to be approximately 43.3% higher than values generated by equilibrium dialysis performed at 37°C, largely because of membrane interactions and differences in filtration dynamics.
Ultracentrifugation as a Membrane-Free Alternative
Ultracentrifugation separates free drug molecules by subjecting plasma samples to extremely high centrifugal forces that sediment plasma proteins into a dense pellet, leaving behind a protein-free supernatant suitable for quantitative analysis. Since this technique does not utilize a semipermeable membrane, it completely avoids membrane-associated non-specific adsorption.
Plasma samples are typically centrifuged at forces exceeding 100,000 × g, with many protocols employing speeds as high as 223,000 × g, for 4 hours or longer while maintaining a temperature of 37°C. Under these conditions, high-density plasma proteins together with their associated drug molecules sediment to the bottom of the centrifuge tube, forming a compact pellet. The resulting clear, protein-depleted supernatant is then carefully removed and analyzed for the unbound drug concentration.
Ultracentrifugation is particularly valuable for compounds that exhibit strong membrane interactions or are considered highly “sticky.” Nevertheless, this methodology has several practical limitations, including relatively low analytical throughput, expensive instrumentation requirements, and the potential formation of sedimentation gradients. These factors may contribute to higher measured free fractions for highly lipophilic compounds compared with standard reference techniques. Studies investigating valproic acid protein binding, for example, have demonstrated that while equilibrium dialysis and ultrafiltration frequently produce closely comparable results, ultracentrifugation may generate divergent values because of incomplete protein sedimentation or back-diffusion phenomena.
Maximize efficiency by leveraging an integrated chemistry and bioanalytical CRO for seamless project execution.
Kinetic Flux Dialysis and Novel SPME Devices
Flux dialysis and Solid-Phase Microextraction (SPME) represent modern kinetic extraction techniques that quantify unbound drug fractions without requiring complete thermodynamic equilibrium. These approaches are particularly advantageous for compounds exhibiting extremely high plasma protein binding (f_u < 0.001) or for analytes that demonstrate extensive non-specific binding.
Flux dialysis operates according to the principle that the initial rate of molecular transport across a dialysis membrane is directly proportional to the product of the initial drug concentration, the unbound fraction, and the membrane permeability coefficient (P_mem).
J0 = Pmem × A × fu × Cinitial
In this system, plasma is placed on both sides of the dialysis membrane, effectively balancing non-specific binding effects. The fu value is subsequently determined from the initial slope of the concentration ratio versus time curve, thereby eliminating the need for the prolonged equilibrium period required by conventional equilibrium dialysis while also reducing challenges associated with matrix instability.
Similarly, the Supel BioSPME 96-pin device employs a non-exhaustive extraction mechanism. Each pin is coated with a proprietary extraction phase that selectively captures small, unbound analytes, while steric hindrance prevents larger macromolecules from interacting with the coating surface. This design enables a rapid, highly efficient workflow for directly measuring free drug fractions in both human and animal plasma. Published studies have demonstrated strong agreement between BioSPME-generated results and measurements obtained using the gold-standard equilibrium dialysis technique.
| Analytical Parameter | Equilibrium Dialysis (ED/RED) | Ultrafiltration (UF) | Ultracentrifugation (UC) | Flux Dialysis | BioSPME (96-Pin Device) |
|---|---|---|---|---|---|
| Physical Principle | Passive membrane diffusion until thermodynamic equilibrium is achieved. | Pressure-driven size exclusion using centrifugation. | Sedimentation of plasma proteins under ultra-high centrifugal force (>100,000 × g). | Kinetic measurement based on the initial membrane flux rate. | Non-exhaustive selective adsorption using a proprietary binder coating. |
| Separation Interface | Semipermeable membrane (typically 8–12 kDa). | Filtration membrane (10–30 kDa). | No membrane; separation occurs through sedimentation. | Semipermeable membrane with plasma on both sides. | Coated solid-phase microextraction pins. |
| Analytical Throughput | High through automated 96-well formats. | Moderate, depending on centrifuge capacity. | Low because of labor-intensive processing. | Moderate to high with scalable workflows. | High using automated 96-pin technology. |
| Workflow Duration | Approximately 4–8 hours. | Approximately 15–30 minutes. | Approximately 4 hours. | Short because equilibrium is not required. | Rapid extraction, generally under 30 minutes. |
| Risk of Adsorption (NSB) | Low to moderate; minimized using inert materials. | High because of membrane and housing interactions. | None due to the absence of a membrane. | Minimal because bilateral plasma minimizes adsorption effects. | Low with highly purified sample extracts. |
| Matrix Stability Impact | High because prolonged incubation at 37°C may degrade unstable compounds. | Low because rapid separation preserves labile analytes. | Moderate because of prolonged centrifugation at 37°C. | Low owing to minimal sample exposure time. | Minimal because of rapid processing at room temperature or 37°C. |
| Regulatory Acceptance | Primary gold-standard method preferred by FDA and EMA. | Secondary approach requiring extensive validation. | Primarily used as a supplemental validation technique. | Increasingly accepted under ICH M12 guidance. | Emerging methodology primarily used during discovery and lead optimization. |
How are Technical Challenges and Artifacts Controlled in Bioanalytical Binding Assays?
Bioanalytical laboratories minimize experimental artifacts such as osmotic volume shifts, non-specific surface adsorption, and pH fluctuations by implementing mathematical correction models, membrane pre-treatment strategies, and controlled carbon dioxide incubation environments. Effectively controlling these analytical variables is essential for producing reproducible, reliable, and regulatory submission-ready free fraction datasets.
Managing Osmotic Volume Shifts
Osmotic volume shifts occur during equilibrium dialysis when water migrates into the hyperosmotic plasma compartment, causing dilution of plasma proteins and artificially increasing the apparent fraction unbound. Because plasma contains a high concentration of non-diffusible macromolecules, primarily albumin, it generates considerably higher oncotic pressure than the aqueous buffer located in the receiver compartment. This osmotic pressure gradient drives water across the semipermeable membrane, resulting in expansion of the plasma volume, typically by 10% to 30%. As plasma proteins become diluted, the number of available drug-binding sites per unit volume decreases, creating the false appearance of an increased free drug fraction.
To compensate for this effect, the post-dialysis fraction unbound can be corrected by comparing the protein concentrations measured before and after dialysis to establish an appropriate correction factor. This approach has been demonstrated in studies involving prednisolone and verapamil, where failure to apply volume corrections produced substantial errors in estimating protein-binding capacity. The corrected fraction unbound (f_u, corrected) is calculated using the following equation:
fu, corrected = Cbuffer × [Protein]post Cplasma, post × [Protein]pre
Where:
- [Protein]pre represents the total plasma protein (or albumin) concentration measured before dialysis.
- [Protein]post represents the total plasma protein (or albumin) concentration measured after dialysis.
As an alternative strategy, osmotic water movement can be minimized by adding an osmotic balancing agent, such as 2.5% (w/v) dextran, to the buffer compartment or by using low-volume, thick-walled Rapid Equilibrium Dialysis (RED) plates specifically designed to reduce water transfer across the membrane.
Learn how to prevent, identify, and troubleshoot bioanalytical method validation failure before it compromises your study.
Resolving Non-Specific Binding and the Recovery Acceptance Myth
Non-specific binding (NSB) to dialysis membranes and device surfaces can substantially reduce analyte recovery. To address this issue, laboratories commonly employ low-adsorption consumables together with membrane pre-treatment using surfactants. Highly lipophilic and basic compounds are especially susceptible to adsorption onto plastic laboratory ware, pipette tips, and regenerated cellulose membranes, which can reduce analyte concentrations below the detection capability of the mass spectrometer. Pretreating dialysis membranes with non-ionic surfactants such as 0.1% Tween-20 or Solutol HS-15, along with the use of low-binding polypropylene consumables, effectively blocks hydrophobic adsorption sites and significantly improves analyte recovery.
However, an important scientific observation described by Pfizer’s Li Di and colleagues introduced what is commonly referred to as the “recovery paradox” in equilibrium dialysis assays. Once complete thermodynamic equilibrium has been established, passive diffusion ensures that the concentration of free drug is identical on both sides of the dialysis membrane. Although adsorption to membranes or device surfaces reduces the total quantity of drug remaining in solution, it does not alter the thermodynamic equilibrium between the bound and unbound drug populations. Consequently, relying on absolute recovery as a strict assay acceptance criterion may be unnecessarily restrictive. Even when recovery is reduced because of non-specific binding, the calculated f_u remains accurate, provided that the remaining analyte concentration within the buffer compartment remains above the analytical instrument’s lower limit of quantification (LLOQ).
Ready for the clinic? Ensure your early-phase trials are supported by expert bioanalytical CRO services for first-in-human studies.
Regulating Plasma pH via Carbon Dioxide Atmosphere
Loss of dissolved carbon dioxide from stored or incubated plasma causes the plasma pH to gradually increase toward 8.7, altering the structural conformation of plasma proteins and introducing significant bias into protein-binding measurements. Under physiological conditions, plasma is naturally buffered by the carbonic acid-bicarbonate (H₂CO₃/HCO₃⁻) buffering system. During incubation at 37°C under ambient atmospheric conditions, dissolved carbon dioxide escapes from the plasma into the surrounding air. This process shifts the bicarbonate equilibrium, depletes dissolved carbon dioxide, and progressively elevates plasma pH.
CARBON DIOXIDE / pH DRIFT IN DIALYSIS
Ambient Air Incubation (Uncontrolled) Controlled CO₂ Atmosphere (Physiological)
CO₂ Degassing CO₂ Maintained at 5%
│ │
▼ ▼
Plasma pH rises to 8.7 Plasma pH maintained at 7.40 ± 0.10
│ │
▼ ▼
Albumin transitions to its basic (B) form Proteins remain in their native physiological
Binding constants become distorted conformation, producing highly accurate and
reproducible free fraction measurements
Human serum albumin undergoes a substantial conformational transition from its normal physiological neutral (N) configuration to the basic (B) configuration when plasma pH exceeds 8.0. This structural alteration changes the geometry of Sudlow’s Site I and Sudlow’s Site II, significantly influencing drug-binding characteristics. For nearly 40% of evaluated compounds, particularly non-polar cationic molecules, increasing the pH from 7.4 to 8.7 produces more than a two-fold change in the measured f_u. To maintain physiological relevance and ensure accurate experimental outcomes, dialysis plates should always be incubated within a humidified incubator operating under a continuous 5% or 10% CO₂ atmosphere, together with a robust 100 mM sodium phosphate buffer adjusted to pH 7.4 in the receiver compartment.
What are the Regulatory Expectations and Guidelines for Protein Binding and Free Fraction Measurement in Bioanalysis?
Regulatory authorities, including the FDA and EMA, expect protein-binding studies to be conducted using fully validated analytical methods that support physiologically relevant clinical pharmacokinetic modeling. Current regulatory expectations are primarily defined within the ICH M10 and ICH M12 guidelines, both of which place considerable emphasis on robust analytical validation whenever experimentally determined unbound drug fractions are incorporated into drug-drug interaction (DDI) risk assessments.
Navigate global submissions with ease: Ensure worldwide compliance by understanding the key EMA vs FDA bioanalytical method validation differences.
The Impact of the ICH M12 Guideline on Sub-Percent Free Fractions
The ICH M12 guideline removes the historical minimum cap of 0.01 for the unbound fraction (f_u), permitting sponsors to use experimentally measured values below 1% in regulatory risk calculations, provided that the analytical methodology has been appropriately validated. Previously, compounds exhibiting extremely high plasma protein binding, for example 99.9% bound (f_u = 0.001), were required to use the default value of f_u = 0.01 when performing static mathematical DDI risk calculations. This conservative assumption originated from the limited sensitivity of earlier-generation mass spectrometers but frequently resulted in substantial overestimation of systemic perpetrator drug concentrations at the liver inlet, ultimately producing exaggerated DDI predictions.
Under the 2024 ICH M12 framework, sponsors may now utilize experimentally determined f_u values below 0.01, significantly improving the accuracy of clinical risk assessments. For example, during evaluation of hepatic uptake transporter inhibition involving OATP1B1 and OATP1B3, the regulatory model estimates the liver inlet R-value according to the following equation:
R = 1 + fu,p × Iin, max IC50
When the historical default value of fu,p = 0.01 is applied to highly protein-bound compounds, the resulting R-value may incorrectly exceed the regulatory threshold, triggering unnecessary clinical DDI studies. Demonstrating acceptable analytical accuracy and precision at sub-percent free fractions enables the use of experimentally measured values, resulting in more realistic predictions and facilitating a smoother regulatory review process.
Ensure your quantitative analyses are fully compliant: Stay up to date with the latest ICH M10 bioanalytical method validation guidelines for global regulatory success.
COMPARATIVE DDI RISK PREDICTIONS: CAPPED VS. MEASURED f_u
Compound / Case Study Applied f_u Predicted R-Value Observed Clinical DDI
---------------------------------------------------------------------------------------------
Itraconazole (CYP3A4) 0.01 (Cap) 10.0 to 30.0-fold 5.0 to 10.8-fold
0.002 5.0 to 10.0-fold Accurate prediction
OATP1B1 Inhibitor 0.01 (Cap) 6.1 1.8-fold AUC shift
0.002 2.0 Highly accurate prediction
Key Bioanalytical Validation Requirements under ICH M10 Principles
Bioanalytical method validation for protein-binding studies requires comprehensive evaluation of accuracy, precision, selectivity, carryover, and matrix effects to satisfy regulatory expectations. When translating biological samples obtained through equilibrium dialysis into LC-MS/MS analysis, the analytical method must be fully qualified according to the principles established within the ICH M10 guideline.
The calibration curve should encompass the entire analytical range, extending from relatively high total plasma concentrations to extremely low sub-nanomolar free drug concentrations measured in the buffer compartment, while maintaining a coefficient of determination of R² ≥ 0.99. Method accuracy and precision must be demonstrated using quality control (QC) samples prepared in multiple lots of the intended biological matrix at four concentration levels:
- Lower Limit of Quantification (LLOQ)
- Low QC
- Medium QC
- High QC
Mean back-calculated concentrations should remain within ±15% of their nominal values, with an allowable deviation of ±20% at the LLOQ. Likewise, the coefficient of variation (CV) for precision should not exceed 15%, with a maximum of 20% permitted at the LLOQ.
ICH M10 VALIDATION PYRAMID
+-----------------------------------------------+
| Linear Calibration Curve (R² ≥ 0.99) |
+-----------------------------------------------+
| QC Accuracy (±15% Bias, LLOQ ±20% Bias) |
+-----------------------------------------------+
| Precision (CV ≤15%, LLOQ CV ≤20%) |
+-----------------------------------------------+
| Matrix Matching & Ion Suppression Controls |
+-----------------------------------------------+
In addition, the post-dialysis matrix-matching procedure must undergo formal validation to demonstrate that adding blank buffer to plasma samples and blank plasma to buffer samples effectively normalizes ionization efficiency during LC-MS/MS analysis. Successful validation confirms complete elimination of matrix-induced ion suppression or ion enhancement, thereby ensuring reliable quantitative determination of free drug concentrations.
Applying the Context-of-Use Validation Framework
The Context-of-Use (CoU) validation framework ensures that bioanalytical assay requirements are scientifically aligned with the intended regulatory purpose of a study, enabling organizations to appropriately balance analytical rigor and resource allocation across both discovery and clinical development programs. During the early stages of drug discovery, screening assays are not expected to comply with formal Good Practice (GxP) requirements. Instead, these assays rely on high-throughput, fit-for-purpose method qualification strategies that rapidly rank candidate compounds according to their fraction unbound (f_u) values and support medicinal chemistry optimization. As development progresses into Investigational New Drug (IND)-enabling studies, clinical drug-drug interaction (DDI) evaluations, and pediatric dose prediction programs, these assays must transition to fully validated methods operating under GxP standards to ensure data integrity, regulatory acceptance, and audit readiness.
Applying a context-driven validation strategy becomes particularly important when evaluating non-traditional therapeutic modalities. Antisense oligonucleotides (ASOs), for example, possess chemically distinct backbone structures that significantly influence their protein-binding behavior. Phosphorodiamidate morpholino oligomers (PMOs) are electrically neutral molecules that generally exhibit relatively low plasma protein binding. In contrast, 2′-O-methoxyethyl/phosphorothioate (MOE/PS)-modified ASOs are highly charged molecules that demonstrate extremely strong and saturable protein binding at concentrations exceeding 1 μM. Validation of free-fraction assays for ASOs commonly incorporates ultrafiltration together with hybridization electrochemiluminescence detection, while dialysis filters are pre-treated with 0.1% Tween-20 to minimize membrane adsorption and achieve the required analyte recovery of greater than 70%.
Developing complex therapeutics? Explore our comprehensive oligonucleotide bioanalytical services tailored specifically to the unique challenges of ASOs and siRNAs.
A similar context-specific validation strategy is necessary for small interfering RNA (siRNA) therapeutics. These molecules possess highly negatively charged, hydrophilic backbones with molecular weights generally ranging between 10 and 15 kDa. Because of their unique physicochemical properties, siRNA therapeutics require hybrid validation approaches that combine principles from both small-molecule and large-molecule bioanalytical guidelines. These studies typically evaluate serum stability, transporter-mediated disposition, and degradation pathways. Likewise, in clinical oncology applications, hydrophilic and metal-sensitive compounds such as intact carboplatin require specialized analytical workflows. In these cases, partial protein precipitation (PPP) combined with Phree phospholipid removal (PPR) is employed to release the analyte from plasma protein binding while avoiding excessive protein precipitation. This approach preserves sample integrity, minimizes matrix interference, and reduces the risk of chromatographic fouling during instrumental analysis.
| Modality Category | Average Molecular Weight | Primary Charge & Hydrophilicity | Primary Plasma Protein Target | Major Validation & Analytical Challenge | Optimized Mitigation Strategy |
|---|---|---|---|---|---|
| Small Molecules | Approximately 0.5 kDa | Hydrophobic; may be acidic, basic, or neutral | Human Serum Albumin (HSA) and α₁-Acid Glycoprotein (AGP) | Extensive non-specific binding (NSB) and lower limit of quantification (LLOQ) sensitivity limitations when f_u < 0.001 | Matrix-matched high-sensitivity LC-MS/MS, flux dialysis, and surfactant membrane pre-treatment |
| Antisense Oligonucleotides (MOE/PS) | Approximately 7 kDa | Highly negatively charged and hydrophilic | Albumin, AGP, and Human γ-Globulins | Saturable protein binding above 1 μM together with extensive membrane adsorption during separation | Ultrafiltration combined with hybridization electrochemiluminescence and 0.1% Tween-20 membrane pre-treatment |
| Antisense Oligonucleotides (PMO) | Approximately 8 kDa | Electrically neutral and hydrophilic | Human γ-Globulins with generally low protein binding | Limited tissue uptake, rapid renal elimination, and relatively poor mass spectrometric ionization efficiency | Hybridization-based analytical methods together with optimization of mobile-phase ion-pairing reagents |
| siRNA Therapeutics | Approximately 10–15 kDa | Highly negatively charged and strongly hydrophilic | Albumin and lipoproteins | Hybrid preclinical validation requirements, rapid serum nuclease degradation, and limited membrane permeability | Chemical modification through GalNAc conjugation, cold extraction procedures, and nuclease-resistant backbone chemistries |
| Monoclonal Antibodies | Approximately 150 kDa | Hydrophilic molecules with complex surface charge distributions | FcRn receptors, target antigens, and non-specific globulin interactions | Molecular size prevents conventional dialysis and may interfere with ligand-binding assays | Size-exclusion chromatography, ligand-binding assays (LBA), or LC-MS/MS following trypsin digestion |
Conclusion on Protein Binding and Free Fraction Measurement in Bioanalysis
Accurate determination of the unbound drug fraction within systemic circulation requires the successful integration of physical chemistry principles, tightly controlled experimental conditions, and thoroughly validated bioanalytical methodologies. By implementing optimized Protein Binding and Free Fraction Measurement in Bioanalysis, drug development programs can move beyond historical assumptions and conservative default values, replacing them with experimentally measured, mathematically validated, and highly sensitive data. This approach improves the accuracy of clinical dose prediction, strengthens pharmacokinetic modeling, enhances drug-drug interaction assessments, and reduces regulatory uncertainty during safety evaluations and marketing submissions.
As a trusted industry partner, ResolveMass Laboratories Inc. provides comprehensive GxP-compliant ADME and bioanalytical services that support pharmaceutical and biotechnology sponsors throughout every stage of drug development, from early discovery through regulatory approval. By integrating advanced mass spectrometry platforms, including the SCIEX MS 6500+, with carefully optimized matrix-matching strategies and rigorous control of pH and temperature conditions, ResolveMass Laboratories Inc. delivers bioanalytical datasets that consistently meet the highest standards for analytical accuracy, precision, robustness, and reproducibility.
To discuss the specific bioanalytical requirements of your development program or obtain expert guidance regarding Protein Binding and Free Fraction Measurement in Bioanalysis, please contact the scientific consulting team through the ResolveMass Laboratories Inc. Contact Us page.
Frequently Asked Questions
Ultrafiltration can overestimate the free fraction because the separation process is influenced by centrifugation conditions, particularly temperature and membrane-related effects. Even slight deviations from the physiological temperature of 37°C may reduce drug-protein binding affinity, allowing more unbound drug to pass through the filtration membrane. In addition, concentration polarization and minor protein accumulation near the membrane can disturb the natural binding equilibrium. As a result, ultrafiltration may produce higher f_u values than equilibrium dialysis, which measures free drug under true thermodynamic equilibrium.
Phosphorodiamidate morpholino oligomers (PMOs) and 2′-O-methoxyethyl/phosphorothioate (MOE/PS) antisense oligonucleotides exhibit markedly different protein-binding characteristics because of their chemical structures. PMOs possess a neutral backbone that interacts minimally with plasma proteins, leading to relatively low protein binding and rapid renal elimination. In contrast, MOE/PS-modified ASOs contain negatively charged phosphorothioate groups that strongly associate with proteins such as albumin and human γ-globulins. This results in extensive, concentration-dependent protein binding and prolonged systemic retention.
During equilibrium dialysis, water can migrate into the plasma compartment because of osmotic pressure differences, diluting plasma proteins and reducing the apparent number of available binding sites. If this dilution is ignored, the calculated free fraction may appear artificially higher than its true physiological value. Measuring protein concentrations both before and after dialysis allows the extent of dilution to be quantified accurately. Applying this correction improves the reliability of protein-binding calculations and produces results that more closely represent the original biological conditions.
The finalized ICH M12 guideline increased the scaling factor used in static CYP induction models from earlier draft recommendations to improve the sensitivity of clinical risk assessments. This modification helps ensure that potent inducers are not overlooked when experimentally measured plasma free fractions (f_u,p) are extremely low. Because highly protein-bound drugs often have sub-percent unbound fractions, the revised scaling approach maintains an appropriate safety margin while allowing validated experimental f_u,p values to be incorporated into regulatory evaluations. This results in more balanced and scientifically supported induction predictions.
Hydrophilic, metal-sensitive compounds such as intact carboplatin may become trapped within dense protein pellets formed during complete protein precipitation using large volumes of organic solvent. This entrapment can substantially reduce analyte recovery and compromise quantitative accuracy. Partial protein precipitation (PPP) uses a milder extraction approach that releases the drug from plasma proteins without causing excessive protein aggregation. When combined with phospholipid removal (PPR), this strategy produces cleaner extracts, improves analytical recovery, and preserves the integrity of the intact analyte.
Radiolabeled protein-binding studies require exceptionally high radiochemical purity because even trace impurities can significantly influence measurements of highly protein-bound compounds. When the true free fraction is below 1%, a small amount of unbound radioactive impurity may diffuse across the dialysis membrane and be mistakenly quantified as free parent drug. This can substantially inflate the calculated f_u and compromise study accuracy. Maintaining radiochemical purity at 99% or higher minimizes analytical interference and ensures reliable determination of the true unbound fraction.
The anticoagulant used during blood collection can affect plasma composition, sample stability, and ionization efficiency during LC-MS/MS analysis. Anticoagulants such as K₂EDTA, sodium heparin, and citrate may influence protein interactions or generate matrix differences that affect analyte recovery. For example, heparinized plasma may occasionally form micro-clots after repeated freeze-thaw cycles, potentially trapping lipophilic compounds. To achieve accurate calibration and minimize matrix effects, the same anticoagulant should be used consistently for study samples, calibration standards, and quality control samples.
Although non-specific binding generally does not alter the equilibrium relationship between bound and free drug, extremely high adsorption to membranes or laboratory surfaces can reduce the remaining free drug concentration below the analytical lower limit of quantification (LLOQ). When the analytical instrument cannot reliably detect these very low concentrations, the measured f_u may be underestimated because of inaccurate baseline measurements or apparent zero values. Improving assay sensitivity through advanced LC-MS/MS instrumentation or optimized sample preparation helps prevent this analytical limitation.
Following extensive review of published evidence and retrospective analyses of regulatory submission data, the finalized ICH M12 guideline concluded that the previous 50-fold scaling factor for reversible CYP inhibition was overly conservative. The revised 5-fold factor provides a more balanced approach by maintaining protection against false-negative predictions while reducing the number of false-positive DDI assessments. Consequently, sponsors can generate more realistic clinical risk projections and avoid unnecessary clinical interaction studies without compromising patient safety.
Reference:
- U.S. Food and Drug Administration. (2024). Drug interaction studies for therapeutic proteins: Study design, data analysis, and clinical implications (Guidance for industry). https://www.fda.gov/media/184437/download
- Liu, Y., Zhang, X., Wang, J., Li, H., Chen, Z., & Zhao, Y. (2025). Characterizing the plasma protein binding profiles of chemistry diversified antisense oligonucleotides in human and mouse plasma using an ultrafiltration method. Frontiers in Pharmacology, 15, Article 1515143. https://doi.org/10.3389/fphar.2024.1515143
- Curran, R. E., Claxton, C. R. J., Hutchison, L., Harradine, P. J., Martin, I. J., & Littlewood, P. (2011). Control and measurement of plasma pH in equilibrium dialysis: Influence on drug plasma protein binding. Drug Metabolism and Disposition, 39(3), 551–557. https://doi.org/10.1124/dmd.110.036988
- Ahmed, H., Bergmann, F., & Zeitlinger, M. (2022). Protein binding in translational antimicrobial development: Focus on interspecies differences. Antibiotics, 11(7), 923. https://doi.org/10.3390/antibiotics11070923
- David, B. M., Tjokrosetio, R., & Ilett, K. F. (1983). Plasma protein binding of disopyramide by equilibrium dialysis and ultrafiltration. Therapeutic Drug Monitoring, 5(1), 81–86. https://doi.org/10.1097/00007691-198303000-00007
- U.S. Food and Drug Administration. (2022). M10 bioanalytical method validation and study sample analysis: Guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m10-bioanalytical-method-validation-and-study-sample-analysis

