
Introduction: Why Risk Allocation Matters More Than Risk Avoidance in Generic Development
In complex generic pharmaceutical development, risk cannot be eliminated. The real challenge is deciding who manages the risk, when it appears, and how responsibilities are shared. CRO Risk Sharing Pharma models have emerged as practical solutions for programs where scientific uncertainty and regulatory variability are expected. When a 505(b)(2) pathway, a complex NDA, or an ANDA for a non-oral dosage form enters development, factors such as bioequivalence uncertainty, formulation complexity, and extended review timelines make traditional fee-for-service models difficult. These realities push sponsors and CROs toward shared accountability structures. In such cases, risk-sharing frameworks help maintain development momentum even when technical or regulatory hurdles arise.
Explore the Essentials of Generic Drug Development: Understanding the Generic Drug Development Process for ANDA
This article focuses on the operational and contractual mechanics that determine whether a risk-sharing arrangement in complex generic development actually works. Poorly structured agreements can unintentionally shift most of the operational burden to one party while only appearing collaborative. Effective models clearly define responsibilities, funding triggers, and decision-making authority in advance. These details help avoid confusion when development timelines change or technical issues occur. Understanding these mechanics is essential before entering any co-development structure involving complex dosage forms or uncertain regulatory pathways.
Share via:
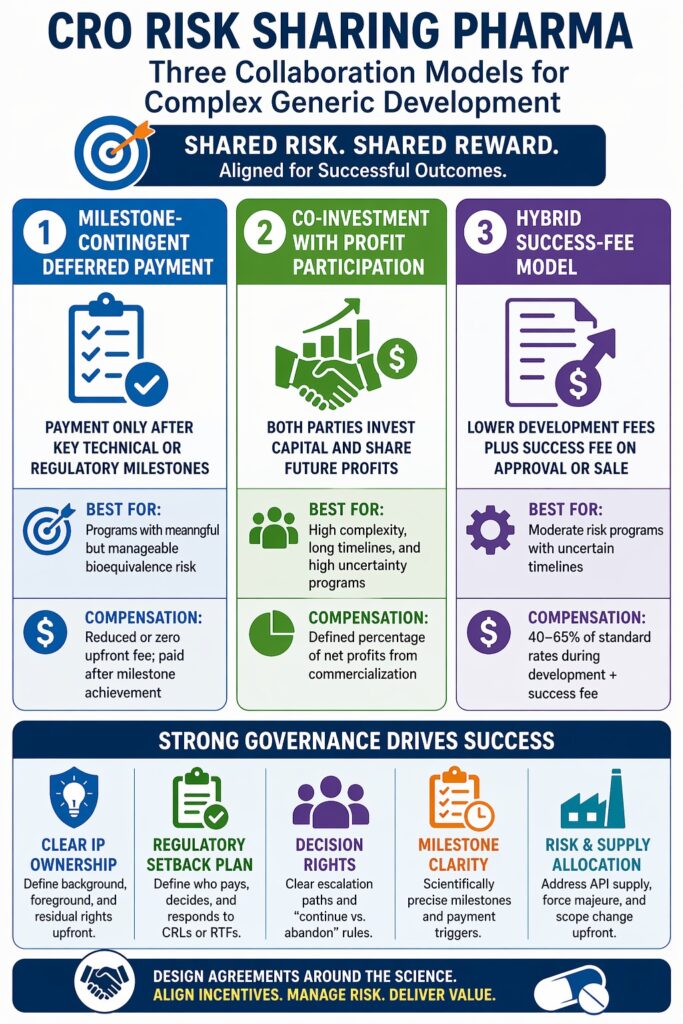
The Three Structural Models of CRO Risk Sharing in Complex Generic Pharma
Risk-sharing in pharma CRO partnerships typically follows three structural approaches: milestone-contingent deferred payment, co-investment with equity or profit participation, and hybrid success-fee models layered onto reduced base fees. Each model distributes scientific, regulatory, and financial exposure differently between sponsor and CRO. The best structure depends on formulation complexity, expected development timelines, and predictability of regulatory expectations. Choosing the wrong model can misalign incentives and create disputes when development changes direction. Therefore, sponsors and CROs should evaluate technical risk before finalizing financial structure.
In-House vs. CRO: Which is Right for Your ANDA? Compare CRO vs. In-House ANDA Development Strategies
1. Milestone-Contingent Deferred Payment
In this model, the CRO performs development work at a significantly reduced or zero upfront fee. Costs and margins are recovered only after predefined technical or regulatory milestones are achieved, such as ANDA acceptance, tentative approval, or commercial launch. This structure shifts early-stage financial exposure to the CRO while linking compensation to measurable progress. It is commonly used for programs where bioequivalence risk is meaningful but manageable. However, the model requires clear milestone definitions to avoid disagreements about whether criteria have been met.
Key contractual variables:
- Definition of “milestone” must be scientifically precise (e.g., BE study success defined as 90% CI within 80–125% for AUC and Cmax, not merely “study completion”)
- Clawback provisions if the sponsor out-licenses the asset post-milestone but before approval
- Responsibility allocation for FDA Complete Response Letters (CRLs) — who pays for re-study costs?
Additional provisions often include timeline buffers for regulatory delays and contingency triggers for formulation redesign. Agreements should also clarify whether milestone payments are cumulative or dependent on continued program progress. Without these safeguards, deferred payment models can create disputes during regulatory setbacks. When structured correctly, they allow both parties to share development risk while maintaining technical rigor.
Navigate the Regulatory Requirements: Essential Requirements for ANDA Submission of Generic Drugs
2. Co-Investment with Profit Participation
In this model, both sponsor and CRO contribute capital to the development program. Instead of charging a development fee, the CRO receives a defined percentage of net profits from commercialization. This structure is suitable for programs with long timelines and high scientific uncertainty, where both parties benefit from long-term upside. Co-investment strengthens collaboration because both organizations become financially tied to product success. However, it requires detailed profit calculation methods and commercialization assumptions.
This model is most appropriate for:
| Product Category | Rationale for Co-Investment Model |
|---|---|
| Inhaled corticosteroids (ICS/LABA combos) | Long development cycle (5–8 years), high device-formulation interaction complexity |
| Topical complex generics (emulsions, gels) | Regulatory science still evolving; outcome uncertainty high |
| Ophthalmic suspensions | PK/PD modeling limitations; animal-to-human extrapolation issues |
| Extended-release oral products with NTI APIs | Narrow therapeutic index adds BE study risk layer |
Co-investment agreements must also define commercialization roles, supply responsibilities, and profit distribution timelines. Without clarity on manufacturing scale-up and market entry strategy, financial participation can become difficult to manage. Exit provisions are also necessary if one party chooses not to fund later-stage activities. These agreements work best when both parties align expectations from the beginning. Proper planning helps prevent misunderstandings during commercialization.
Accelerate Your Development Timeline: How CDMOs Accelerate Generic Drug Development in the US and Canada
3. Hybrid Success-Fee Models in CRO Risk Sharing Pharma
The most commonly used CRO Risk Sharing Pharma structure involves the CRO charging 40–65% of standard FTE rates during development, with the remaining portion recovered as a success fee upon ANDA approval or first commercial sale. This approach balances upfront affordability with outcome-based compensation. It is simpler than co-investment because it avoids complex profit accounting. At the same time, it maintains alignment between sponsor and CRO. Hybrid models are often used when development risk is moderate but timelines remain uncertain.
This model requires careful structuring of:
- Rate-lock provisions: Protecting CRO from inflation in personnel costs over multi-year programs
- Scope-change triggers: Preventing sponsor from expanding development scope without renegotiating success-fee terms
- Force majeure clauses: Addressing API shortages, manufacturing disruptions, or FDA review delays
Additional considerations include success-fee payment timing and handling of partial approvals. Agreements should also address situations where approval is achieved but commercialization is delayed. Without these provisions, success-fee recovery may become uncertain. Properly designed hybrid models provide flexibility while maintaining shared accountability.
CRO Risk Sharing Pharma — Where Governance Design Determines Success
The most reliable predictor of success in CRO Risk Sharing Pharma arrangements is governance quality rather than fee structure. Many risk-share failures in complex generic development arise from unclear decision rights, misaligned escalation paths, or undefined IP ownership terms. Scientific setbacks are expected in complex generics, but governance gaps create avoidable disputes. A well-defined governance framework ensures decisions during setbacks are timely and consistent. This structure also preserves collaboration when timelines extend.
IP Ownership in Jointly Developed Formulations
When a CRO contributes proprietary excipient knowledge, particle engineering techniques, or coating technology to a jointly developed generic, IP ownership becomes commercially sensitive. This is especially important if the product fails FDA review and the sponsor decides to discontinue development. Without clear IP definitions, both parties may claim rights to formulation innovations. Such disputes can delay future development opportunities. Therefore, IP governance should be established before experimental work begins.
Critical IP provisions that must be addressed upfront:
- Background IP: Technology each party brings to the collaboration (pre-existing, owned separately)
- Foreground IP: Jointly developed innovations created during the program
- Residual rights: What the CRO may retain and use in future programs if the collaboration terminates
Failing to distinguish between background and foreground IP is one of the most common governance failures observed in stalled co-development programs. Agreements should also define licensing rights if the collaboration ends early. Documentation standards for inventive contributions should also be specified. These measures reduce ambiguity during program transitions.
Regulatory Setback Allocation
In complex generic development, an FDA Refuse-to-File (RTF) or Complete Response Letter is rarely a final failure. Instead, it represents a technical setback requiring defined remediation steps. Risk-sharing agreements must specify how such scenarios are handled financially and operationally. Without clear allocation, each party may hesitate to fund additional studies. This uncertainty can delay remediation timelines.
Risk-sharing agreements must address:
- Who funds the BE re-study if the initial study fails on statistical grounds unrelated to formulation performance?
- Who owns the regulatory correspondence and response strategy if a CRL is issued?
- What is the decision timeline for “continue vs. abandon” and which party holds veto rights?
A well-structured partnership assigns decision rights clearly and ties financial consequences to decisions rather than outcomes. This reduces disputes during regulatory delays. It also ensures that technical decisions remain data-driven. Clear escalation pathways further improve program continuity.
Technical Complexity as a Risk Amplifier — Specific Formulation Challenges
Complex generic formulations increase risk in co-development models because failure modes are difficult to predict and costly to fix. Unlike conventional oral solids, these products often involve device interactions, physicochemical variability, or patient-dependent performance. As a result, CRO Risk Sharing Pharma structures must reflect formulation-specific challenges. Ignoring these technical realities can destabilize partnerships. Contractual terms should align with formulation risk profile.
Strategic Peptide Development: Specialized Peptide CDMO Services for Complex Generics
Pulmonary Drug Products (MDIs, DPIs)
Device-formulation interaction introduces a unique risk layer, as formulation performance cannot be separated from the inhaler device. Variability in airflow resistance, emitted dose, and particle deposition complicates development. Scale-up may also alter aerodynamic particle size distribution. These factors make pulmonary products particularly sensitive to manufacturing changes.
A CRO taking risk on a DPI development program must contractually address:
- Device sourcing and exclusivity risk
- In-vitro/in-vivo correlation (IVIVC) acceptance by FDA
- Particle size distribution reproducibility across manufacturing scale-up
Additional considerations include device lifecycle management and supplier qualification. Agreements should also address equivalence testing across multiple device batches. Without these controls, regulatory acceptance may be affected. Properly structured contracts help reduce these risks.
Transdermal Systems
Membrane-controlled patches and matrix systems face adhesive variability across skin types, differing adhesion standards by market, and crystallization risks during shelf life. These factors can influence drug release and bioequivalence outcomes. Environmental conditions during storage also affect patch performance. As a result, stability becomes a key risk driver.
Risk-sharing agreements for transdermal programs should include formulation stability milestones, not just bioequivalence milestones. Adhesion testing protocols should also be predefined. Agreements should define acceptance criteria for cold-flow and edge-lift behavior. These measures ensure that technical risks are addressed early. Proper planning reduces late-stage surprises.
Liposomal and Nanoparticulate Injectables
FDA guidance for complex injectable generics requires extensive physicochemical characterization. Attributes such as particle size, lamellarity, and encapsulation efficiency must be tightly controlled. Small variations can alter therapeutic equivalence. Consequently, development costs and analytical requirements are higher.
The risk of a “not therapeutically equivalent” determination is structurally higher for liposomal products. Co-development agreements must therefore define acceptable characterization data before advancing to in-vivo studies. This reduces the risk of costly clinical failures. Analytical method ownership should also be clarified. These provisions strengthen risk-sharing effectiveness.
Master the Regulatory Pathway: Understanding the Regulatory Pathway for Complex Peptide Injectables
Structuring Co-Development Agreements That Hold Under Regulatory Pressure
A risk-sharing agreement that has not been tested against realistic regulatory failure scenarios often leads to disputes later. Agreements must anticipate CRLs, study failures, and supply disruptions. Each provision should address a specific operational risk. Without stress-testing, contractual language may remain ineffective. Proper structuring improves program resilience.
| Agreement Provision | Risk Addressed | Common Drafting Failure |
|---|---|---|
| Milestone definition clause | Ambiguous success criteria | Milestones defined by activity completion, not scientific outcomes |
| IP assignment schedule | Foreground IP ownership disputes | Failure to define “inventive contribution” thresholds |
| Regulatory escalation protocol | CRL/RTF response delays | No decision timeline with financial consequences |
| Termination for convenience terms | Sponsor exits program post-milestone | No clawback or compensation floor for CRO |
| API sourcing responsibility | Supply chain disruption delays | Sole-source API risk unallocated |
| Audit and data access rights | Cost verification in deferred models | CRO books inaccessible to sponsor during program |
| Change-of-control provisions | M&A activity affecting either party | Agreement silent on assignment rights post-acquisition |
ResolveMass Laboratories Inc. embeds a pre-execution agreement stress-test into every co-development partnership. This process evaluates multiple realistic failure scenarios. The objective is to ensure that contractual language remains operational under pressure. Such preparation reduces disputes during development.
Evaluating CRO Capability for Risk Sharing — What Sponsors Often Miss
In CRO Risk Sharing Pharma models, willingness to share risk is less important than the ability to manage technical variables. Sponsors must evaluate whether the CRO has experience with complex dosage forms. Operational capability directly influences risk exposure. Without adequate technical depth, risk-sharing becomes difficult to sustain. Therefore, careful due diligence is essential.
Find the Right Partner: Choosing the Best CRO for Leuprolide Depot Development
Sponsors evaluating potential partners should assess:
- Regulatory track record on complex dosage forms: Not ANDA count, but first-cycle approval rates and CRL frequency
- In-house bioanalytical capability: Outsourced bioanalysis introduces third-party dependency
- Formulation science depth: Product-category-specific expertise versus generalist chemistry support
- API characterization capability: Control of polymorphism, particle engineering, and co-crystal development
- Regulatory writing quality: Experience with Category 1 product responses
Additional evaluation should include project management structure and communication cadence. Sponsors should also review historical timelines for similar products. These factors influence execution reliability. Capability assessment ensures alignment before risk sharing begins.
Leverage Proven CMC Strategies: Establishing a Robust Leuprolide Depot CMC Strategy
Conclusion: Designing Risk-Share Models That Reflect the Science, Not Just the Finance
CRO Risk Sharing Pharma models for complex generic development succeed when structured around technical complexity rather than financial negotiation. Delivery system characteristics, API behavior, regulatory pathway, and development timeline should shape risk allocation. Financial structures should then align with these scientific realities. This approach reduces misalignment during setbacks. It also improves long-term collaboration.
At ResolveMass Laboratories Inc., co-development partnerships are structured with technical accountability at their core. Integrated formulation science, regulatory affairs, and bioanalytical capabilities allow shared risk to be actively managed. This ensures that both parties influence outcomes rather than simply sharing financial exposure. Agreements are designed to reflect operational responsibilities at each stage. Such alignment strengthens partnership performance.
If your organization is evaluating risk-sharing structures for a complex generic development program, we invite you to engage our team in a technical and contractual pre-assessment before any agreement is executed. Early evaluation helps identify risks before they affect timelines. It also improves agreement clarity. Collaborative planning supports successful development.
Contact ResolveMass Laboratories Inc.
Frequently Asked Questions (FAQs)
In a CRO Risk Sharing Pharma agreement, milestones should be based on measurable scientific results rather than simple task completion. For example, a milestone may require specific pharmacokinetic values within regulatory limits instead of just finishing a study. If a disagreement occurs, contracts usually refer the issue to a technical review committee with a fixed decision timeline. Disputes often arise when results are partially successful, so agreements should also explain how conditional payments or corrective steps will be handled.
Co-investment models work best for complex generics that require long development timelines and involve higher regulatory uncertainty. Examples include inhaled combination products, ophthalmic suspensions, liposomal injectables, and topical formulations requiring skin permeation studies. These products usually demand significant investment and may face unpredictable approval outcomes. Because of the potential commercial upside, profit-sharing structures can make the collaboration more balanced for both sponsor and CRO.
API sourcing responsibility should always be clearly defined in the contract to avoid delays and disputes. If the sponsor controls API supply, any delays caused by shortages should pause milestone timelines without penalizing the CRO. When the CRO manages API sourcing, qualification milestones and backup supplier plans should be included. This ensures development can continue even if the primary API source fails.
IP ownership depends on how the agreement separates background and newly developed technologies. A CRO typically retains background IP such as formulation platforms, analytical methods, and internal process knowledge. Foreground IP created during the collaboration is usually governed by negotiated terms between both parties. In many cases, the CRO may keep limited rights to reuse certain innovations unless exclusivity has been clearly defined.
Well-structured CRO Risk Sharing Pharma agreements assign responsibility based on the type of deficiency in the CRL. Formulation or analytical issues are usually handled by the CRO, while labeling or regulatory strategy matters remain with the sponsor. Cost allocation for additional studies should also be defined in advance. This is important because CRL remediation for complex generics may require expensive new studies.
Outsourcing bioanalytical work can create gaps in accountability within a risk-sharing model. Bioanalytical method validation and sample analysis directly impact bioequivalence outcomes. When these services are outsourced, an additional third party becomes part of the risk chain. Sponsors should review whether the CRO maintains oversight, liability coverage, and strong contractual controls over outsourced bioanalytical work.
In a hybrid success-fee model, the CRO receives reduced payments during development and the remaining amount after approval or launch. This provides ongoing cash flow while still aligning incentives with program success. In a pure milestone-deferred model, the CRO receives payment only after achieving defined milestones. The hybrid approach is usually more practical for multi-year programs, while the deferred model requires strong financial capacity from the CRO.
Audit rights allow sponsors to verify costs in deferred-fee arrangements. Agreements should permit access to time records, vendor invoices, consumable expenses, and equipment usage data. These audits are typically limited to reasonable notice periods and conducted once per year. Clear timelines for resolving audit findings help prevent delays in development activities.
Reference:
- Stern, S., Coghlan, J., Krishnan, V., Raney, S. G., Babiskin, A., Jiang, W., Lionberger, R., Xu, X., Schwendeman, A., & Polli, J. E. (2021). Research and education needs for complex generics. Pharmaceutical Research, 38(12), 1991–2001. https://doi.org/10.1007/s11095-021-03149-y
- U.S. Food and Drug Administration. (2024). Control of nitrosamine impurities in human drugs: Guidance for industry. https://www.fda.gov/media/188024/download
- Sreedevi, A., Kumari, K., & Rao, Y. M. (2024). A deep dive into the development of complex generics: A comprehensive review. Journal of Applied Pharmaceutical Science, 14(10), 1–14. https://japsonline.com/admin/php/uploads/4400_pdf.pdf

