
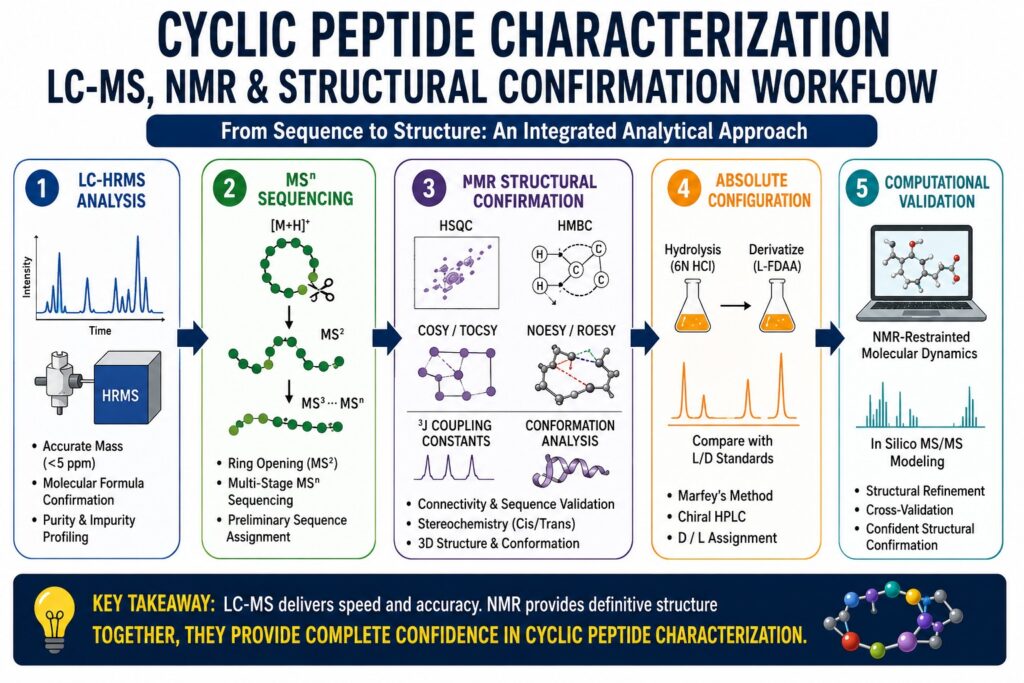
Introduction: Why Cyclic Peptide Characterization Requires a Distinct Analytical Framework
Characterizing cyclic peptides is far more than a straightforward extension of linear peptide analysis. It represents a specialized analytical discipline that demands significant adaptation of established methodologies in mass spectrometry, chromatography, and nuclear magnetic resonance (NMR) spectroscopy. Unlike linear peptides, cyclic peptides lack free N- and C-termini, which serve as critical reference points in conventional sequencing approaches such as Edman degradation. The cyclic architecture also produces highly complex fragmentation patterns in mass spectrometry and introduces conformational constraints that can obscure key residue-to-residue relationships in standard NMR analyses.
As cyclic peptides continue their transition from natural product discovery into pharmaceutical development, the need for reliable and regulatory-compliant characterization workflows has become increasingly important. More than 40 cyclic peptide therapeutics are currently used clinically, including cyclosporin A, vancomycin, and daptomycin. Consequently, analytical laboratories are under growing pressure to establish characterization strategies that are rapid, reproducible, and capable of supporting regulatory submissions. This article explores the analytical methodologies, instrument platforms, fragmentation mechanisms, NMR techniques, and integrated workflows that currently represent best practices for cyclic peptide structural confirmation.
For comprehensive support meeting regulatory standards, explore the ResolveMass Regulatory Requirements for GLP-1 Peptide Characterization.
Key Takeaways
- Comprehensive characterization of cyclic peptides cannot be achieved with a single analytical method; multiple complementary techniques are required to establish structure with confidence.
- High-resolution LC-MS serves as the primary analytical platform for verifying molecular weight, evaluating sample purity, and detecting as well as identifying process- and degradation-related impurities.
- Interpreting MS/MS data for cyclic peptides is significantly more challenging than for linear peptides because ring opening can occur at several locations, generating complex and overlapping fragment ion patterns.
- Advanced NMR techniques, including COSY, TOCSY, HMBC, and NOESY, provide definitive evidence for residue connectivity, stereochemical arrangement, and solution-phase three-dimensional structure.
- Determining the absolute configuration of D-amino acids and non-proteinogenic residues typically relies on Marfey’s method in combination with chiral HPLC analysis.
- The most reliable approach for structural verification combines LC-MS and NMR data with computational modeling and data interpretation tools, creating a robust workflow suitable for research and regulatory applications.
- Unique structural characteristics of cyclic peptides, such as the absence of terminal groups, incorporation of unusual amino acids, and dynamic conformational behavior, require specialized analytical methodologies designed specifically for cyclic peptide analysis.
LC-MS Strategies for Cyclic Peptide Characterization: From Mass Confirmation to Sequence Readout
Liquid chromatography-mass spectrometry (LC-MS) serves as the primary analytical platform for cyclic peptide characterization. It provides confirmation of molecular mass, evaluates chromatographic purity, and generates the initial fragmentation information required for sequence determination. However, successful characterization requires analytical strategies specifically designed for cyclic structures rather than direct application of workflows developed for linear peptides.
Learn how advanced mass spectrometry platforms optimize analysis by viewing the ResolveMass CRO for GLP-1 Peptide Characterization Services.
Choosing the Appropriate Ionization Method and Instrument Platform
For cyclic peptide analysis, electrospray ionization (ESI) is generally favored over matrix-assisted laser desorption/ionization (MALDI) when LC coupling is required. ESI produces multiply charged ions that generate more informative MS/MS spectra and seamlessly integrates with online chromatographic separations. Nevertheless, each mass spectrometry platform presents distinct advantages and limitations.
| Instrument Type | Primary Advantage for Cyclic Peptides | Limitation |
|---|---|---|
| Triple Quadrupole (QqQ) | Excellent sensitivity for targeted MRM quantification | Limited resolution and inability to differentiate isobaric impurities |
| Q-TOF / HRMS | Accurate mass measurement (<5 ppm) and molecular formula confirmation | Higher operational cost per sample |
| Ion Trap (IT) | Supports multi-stage MSⁿ fragmentation essential for ring-opening sequencing | Lower mass accuracy and resolution |
| Orbitrap (HRMS) | Ultra-high resolution, MSⁿ capability, and metabolite profiling | More complex data processing requirements |
For structural characterization at a regulatory standard, LC-HRMS platforms such as Q-TOF and Orbitrap instruments are generally preferred. Their ability to provide accurate mass measurements enables molecular formula determination, differentiation of co-eluting compounds with identical nominal masses, and confident assignment of structurally meaningful fragment ions.
Review specialized workflows for complex architectures via the ResolveMass LC-MS Characterization of GLP-1 Peptides Project.
The Central Challenge: MS/MS Fragmentation of Cyclic Peptides
One of the most technically challenging aspects of cyclic peptide characterization is interpreting tandem mass spectrometry data. During collision-induced dissociation (CID) or higher-energy collisional dissociation (HCD), a protonated cyclic peptide must first undergo cleavage at a random amide bond to generate a linear acylium intermediate. Because ring opening can occur at multiple positions, a collection of isomeric acylium ions with identical m/z values is produced.
As a result, the MS/MS spectrum becomes a composite of numerous overlapping linear fragmentation pathways. This situation contrasts sharply with linear peptides, where fragmentation follows a predictable orientation from the N- and C-termini, producing relatively straightforward b- and y-ion ladders.
Several practical consequences arise from this random ring-opening process:
- Any amino acid residue may effectively become the “N-terminus” of the transient linearized structure.
- A cyclic peptide containing N residues can theoretically generate as many as N independent b-ion series simultaneously.
- Internal fragment ions and satellite ions significantly increase spectral complexity.
- Non-proteinogenic amino acids introduce mass differences that are absent from conventional peptide databases.
Multi-Stage MSⁿ: A Dedicated Sequencing Strategy for Cyclic Peptides
To overcome the ambiguity associated with random ring-opening, multi-stage MSⁿ analysis using ion-trap mass spectrometers has become a highly effective sequencing strategy. This methodology has been extensively validated using NMR-confirmed reference structures and typically follows the workflow outlined below:
MS¹: Detection of the intact cyclic peptide as [M+H]⁺ or multiply charged precursor ions.
MS²: CID fragmentation of the precursor ion generates a population of ring-opened acylium intermediates.
MS³: A selected acylium ion from the MS² spectrum is isolated and subjected to an additional round of CID. This process converts the previously ambiguous cyclic fragmentation into a directional linear fragmentation pathway, progressively removing amino acid residues from the C-terminus until a b₂ ion is produced.
Sequence Assignment: Differences in mass between successive MSⁿ fragments directly correspond to individual amino acid residue masses, enabling sequence determination.
This strategy effectively eliminates the combinatorial complexity inherent in the MS² spectrum by establishing a defined fragmentation starting point. The approach has been successfully applied to cyclic peptides ranging from tetrapeptides to octapeptides, including structures containing non-standard amino acids, and consistently produces sequence assignments that agree with NMR-derived structural data.
Optimize your step-by-step sequencing workflows with the ResolveMass GLP-1 Analog Peptide Sequencing Workflow Guide.
LC Method Development for Purity Assessment and Impurity Profiling
Cyclic peptides present unique chromatographic challenges. Compared with linear peptides, they often display increased hydrophobicity due to the absence of free termini, may exist in multiple conformational states that broaden or split chromatographic peaks, and frequently co-elute with linear precursors or partially cyclized intermediates that possess identical molecular masses.
Important method development considerations include:
- Column Chemistry: Reversed-phase C18 columns with pore sizes between 100 and 130 Å, combined with UPLC/UHPLC particle sizes of 1.7–1.8 µm, generally provide optimal separation efficiency. Phenyl-hexyl stationary phases may offer enhanced resolution of highly hydrophobic cyclic peptide variants.
- Mobile Phase: Common systems employ 0.1% formic acid or 0.1% TFA in water/acetonitrile mixtures. Although TFA can improve chromatographic performance, it suppresses ionization efficiency and is therefore less suitable for LC-MS applications. Ammonium formate or ammonium acetate buffers (10–20 mM) can improve peak shape, particularly for cyclic peptides rich in basic amino acid residues.
- Gradient Design: Shallow gradients of approximately 0.5–1% organic solvent per minute within the critical elution region often improve separation of cyclic peptides from closely related linear isomers and impurity species.
- Column Temperature: Elevated temperatures between 40°C and 60°C help reduce peak broadening caused by conformational exchange processes.
A significant advantage of LC-HRMS is its ability to simultaneously identify and quantify impurities that may be unresolved by UV detection alone. Closely related variants such as deshydro analogs, epimers, and partially open cyclic species may appear as a single chromatographic peak under UV monitoring but can be distinguished through accurate mass measurements.
For reliable regulatory identification of side-products, see the ResolveMass GLP-1 Peptide Impurity Characterization Services.
Critical Consideration for Cyclic and Linear Purity Assessment:
Cyclic and linear forms of the same peptide often possess identical nominal masses when cyclization occurs through head-to-tail amide bond formation. In such cases, conventional mass measurements may be insufficient. Energy-resolved MS/MS approaches, which evaluate fragmentation behavior as a function of collision energy, have emerged as powerful tools for distinguishing cyclic and linear isomers based on their unique fragmentation energy profiles.
NMR Strategies for Cyclic Peptide Structural Confirmation
While mass spectrometry provides molecular weight and sequence information, NMR delivers definitive evidence regarding connectivity, stereochemistry, and three-dimensional structure in solution. Comprehensive characterization of cyclic peptides relies on a coordinated series of one-dimensional and two-dimensional NMR experiments, each addressing specific structural questions.
The NMR Experiment Suite for Cyclic Peptide Characterization
| NMR Experiment | Structural Information Obtained | Specific Role in Cyclic Peptides |
| ¹H NMR (1D) | Chemical shifts, multiplicity, integration | Initial proton inventory; identification of unusual shifts caused by ring-current effects or N-methylation |
| ¹H-¹H COSY | Through-bond connectivity (2–3 bonds) | Identification of amino acid spin systems and Hα–HN relationships |
| TOCSY | Extended spin-system correlations | Assignment of complete residue spin systems, especially in Pro-rich sequences |
| ¹³C HSQC | Direct ¹H-¹³C one-bond correlations | Carbon assignments and identification of N-methylated residues |
| HMBC | Long-range ¹H-¹³C correlations (2–4 bonds) | Confirmation of inter-residue connectivity and cyclization |
| NOESY / ROESY | Through-space proton interactions (<5 Å) | Conformation analysis and stereochemical determination |
| DQF-COSY | Enhanced J-coupling resolution | Precise measurement of vicinal coupling constants |
Discover how multi-dimensional NMR can validate your peptide structure through ResolveMass 2D NMR for Peptide Characterization.
Establishing Cyclic Connectivity Through HMBC
Among all NMR experiments, HMBC is arguably the most important for confirming cyclic peptide architecture. Because cyclic peptides lack terminal groups, the ring-closing amide bond cannot be validated using COSY or HSQC alone. HMBC overcomes this limitation by detecting long-range proton-carbon correlations spanning two to four bonds.
These correlations allow direct observation of relationships between NH or Hα protons and carbonyl carbons across amide linkages. Such connectivity patterns provide compelling evidence that the peptide exists in a cyclic rather than linear form.
Key HMBC correlations include:
- NH of residue i → carbonyl carbon of residue i−1.
- Hα of residue i → carbonyl carbon of residue i.
- Hα of residue i → carbonyl carbon of residue i−1.
- Side-chain protons → carbonyl carbons involved in non-standard linkages such as ester bonds or branched cyclizations.
Particular attention must be given to confirming the ring-closing bond, as its presence provides definitive evidence of successful cyclization.
Three-Dimensional Conformation Through NOESY and Coupling Constant Analysis
Once cyclic connectivity has been established, NOESY and ROESY experiments provide insight into solution-phase conformation. The Nuclear Overhauser Effect measures spatial proximity between protons, with cross-peak intensity depending strongly on internuclear distance.
For cyclic peptides, these experiments reveal two critical structural features.
1. Cis/Trans Amide Bond Configuration
Trans amide bonds typically display strong Hα(i)–HN(i+1) NOE interactions and weak Hα(i)–Hα(i+1) correlations. In contrast, cis amide bonds produce characteristic Hα(i)–Hα(i+1) NOEs.
Because cis conformations occur far more frequently in cyclic peptides, particularly those containing proline or N-methylated residues, accurate assignment of amide geometry is essential. A single incorrect assignment can compromise the entire conformational model.
2. Dihedral Angle Constraints from ³J Coupling Constants
Vicinal coupling constants (³J_HNHα), measured using DQF-COSY or extracted from high-resolution ¹H spectra, can be converted into φ dihedral angle constraints through application of the Karplus equation.
When combined with NOESY-derived distance restraints, these data provide the structural constraints necessary for NMR-restrained molecular dynamics simulations and conformational ensemble generation.
Recent developments from 2024–2025 have demonstrated that integrating traditional isotropic NMR parameters with anisotropic measurements such as Residual Dipolar Couplings (RDCs) allows determination of flexible cyclic peptide conformations with accuracy approaching DFT-level structural models.
Learn how optical techniques complement conformational studies by reviewing ResolveMass CD Spectroscopy for Peptide Secondary Structure Characterization.
Absolute Configuration: Marfey’s Method and Chiral Analysis
Neither NMR nor MS can independently distinguish L- and D-amino acid residues. Therefore, determination of absolute configuration requires dedicated chiral analytical methods.
Marfey’s Method:
The cyclic peptide is completely hydrolyzed using 6N HCl at 110°C for 24 hours. The liberated amino acids are then derivatized with L-FDAA (1-fluoro-2,4-dinitrophenyl-5-L-alanine amide). The resulting diastereomeric derivatives are separated by RP-HPLC and compared against authentic standards. Marfey’s method remains the gold standard for absolute configuration determination in cyclic peptides.
Chiral HPLC:
Direct separation of amino acids on chiral stationary phases provides an alternative approach, particularly when derivatized products exhibit overlapping retention times.
Optical Rotation and ECD:
These techniques serve as complementary tools for assessing overall molecular chirality. Although Electronic Circular Dichroism can provide information regarding global stereochemistry, it generally cannot assign configuration at individual amino acid residues.
Integrated Characterization Workflows: Combining LC-MS and NMR for Definitive Structural Confirmation
Definitive cyclic peptide characterization requires a sequential workflow in which information from mass spectrometry informs NMR analysis, while NMR data validates sequence proposals generated from MS experiments. Neither technique alone provides the level of structural certainty necessary for novel cyclic peptide characterization.
Recommended Workflow for New or Complex Cyclic Peptides
Step 1: LC-HRMS Analysis
- Accurate molecular mass determination using ESI-HRMS.
- Molecular formula calculation with mass accuracy below 5 ppm.
- Purity assessment using UV detection and total ion chromatograms.
- Preliminary impurity identification through accurate mass measurements.
Step 2: MS/MS Sequencing
- CID-mediated ring-opening fragmentation.
- Multi-stage MSⁿ sequencing in ion-trap systems.
- Generation of preliminary sequence assignments.
Step 3: NMR Structural Confirmation
- ¹H NMR for proton assignment.
- HSQC and HMBC for connectivity validation.
- COSY and TOCSY for spin-system identification.
- NOESY and ROESY for conformational analysis.
- ³J coupling constant measurements for dihedral angle determination.
Step 4: Absolute Configuration Analysis
- Marfey’s method for D/L assignment.
- Chiral HPLC as a confirmatory technique.
Step 5: Computational Validation
- NMR-restrained molecular dynamics simulations.
- In silico MS/MS fragmentation modeling.
When LC-MS Alone Is Adequate and When NMR Becomes Essential
For well-established cyclic peptide APIs with validated reference spectra, LC-HRMS combined with impurity profiling may be sufficient for routine quality control, stability studies, and batch release testing.
However, NMR remains indispensable in the following situations:
- Initial structural characterization of a newly discovered cyclic peptide.
- Differentiation of constitutional isomers with identical masses.
- Verification of cyclization regiochemistry.
- Assignment of absolute configuration in non-standard residues.
- Resolution of ambiguous MSⁿ sequence assignments involving isobaric amino acids.
Key Insight:
Leucine and isoleucine possess identical residue masses (113.084 Da) and are therefore indistinguishable by mass spectrometry alone. In cyclic peptides containing these residues, NMR techniques such as NOESY and HMBC provide the most reliable non-derivatization approach for differentiation.
For industry-validated verification frameworks, review the ResolveMass Peptide Sameness Study for ANDA Applications.
Special Structural Challenges in Cyclic Peptide Characterization
N-Methylated Residues and Non-Proteinogenic Amino Acids
N-methylated amino acids, common in nonribosomal peptides such as cyclosporin A, introduce several analytical challenges.
- NMR: Absence of NH resonances necessitates identification through characteristic N-methyl carbon signals observed in HSQC and confirmation through HMBC connectivity.
- MS/MS: N-methylation introduces a 14 Da mass increase that must be incorporated into sequence interpretation workflows.
- Conformation: These residues strongly favor cis amide geometries, which require direct NOESY confirmation.
Disulfide-Bridged Cyclic Peptides
Cyclic peptides stabilized through disulfide bonds require specialized analytical approaches.
Reduction and Alkylation for MS Analysis:
Treatment with DTT followed by iodoacetamide alkylation converts disulfide bonds into stable carbamidomethyl derivatives, enabling straightforward MS/MS sequencing.
Oxidized versus Reduced Species:
LC-MS should quantify both forms because incomplete cyclization may represent a critical quality attribute.
NMR Characterization:
NOESY experiments conducted on the oxidized peptide provide direct evidence of the disulfide-constrained conformation. Characteristic downfield shifts of cysteine Hβ protons further support disulfide bond formation.
Cyclodepsipeptides and Ester Bond Identification
Cyclodepsipeptides contain one or more ester bonds in addition to amide linkages. Because ester bonds possess lower bond dissociation energies, they fragment preferentially during MS/MS analysis, generating characteristic fragmentation patterns.
In NMR spectra, ester carbonyl carbons typically resonate slightly downfield relative to amide carbonyls. However, the most reliable diagnostic feature is the absence of an NH proton at the ester linkage position, confirmed through HMBC correlations between the hydroxy acid α-proton and the ester carbonyl carbon.
Software and Data Processing Tools for Cyclic Peptide MS/MS Interpretation
Manual interpretation of cyclic peptide MS/MS spectra can be labor-intensive and susceptible to error, especially for large or structurally unusual molecules. Several software platforms have been developed to assist with data interpretation.
| Tool | Function | Key Feature |
| mMass | Cyclic peptide MS/MS annotation | Open-source platform supporting non-proteinogenic monomers |
| MS-CPA (UCSD) | Web-based tandem MS annotation | Designed specifically for nonribosomal cyclic peptides |
| GNPS (Global Natural Products Social) | Molecular networking | Dereplication of cyclic natural products through spectral similarity |
| UNIFI (Waters) | LC-HRMS data processing | Compliance-ready pharmaceutical workflow |
| Skyline | MRM method development | Quantitative targeted analysis of known cyclic peptides |
Conclusion: Building a Robust Cyclic Peptide Characterization Strategy
Achieving the level of certainty required in modern drug development and natural products research demands a comprehensive, multi-technique approach to cyclic peptide characterization. LC-HRMS provides the speed, sensitivity, and mass accuracy necessary for molecular weight confirmation, impurity profiling, and purity assessment. Multi-stage MSⁿ sequencing addresses the unique challenges associated with cyclic backbone fragmentation and sequence determination. NMR contributes the critical structural information that mass spectrometry alone cannot provide, including definitive connectivity, stereochemistry, and solution-state conformation.
Through the combined application of HMBC for connectivity analysis, NOESY for conformational characterization, and coupling constant analysis for dihedral angle determination, NMR establishes the structural framework of the cyclic peptide. When integrated with Marfey’s method, the complete stereochemical composition of the molecule can also be determined.
The workflows presented in this article reflect contemporary best practices supported by peer-reviewed research and are applicable to a broad range of cyclic peptide classes, including synthetic drug candidates, nonribosomal natural products, and constrained therapeutic peptides.
Secure regional and international project compliance by utilizing either ResolveMass Peptide Sameness Study Services in Canada
or ResolveMass Peptide Sameness Study Services in the United States.
For research groups confronting the analytical challenges associated with novel cyclic peptides, whether for initial structural elucidation, regulatory impurity profiling, or resolution of complex MS/MS data, the most reliable strategy remains an integrated and hypothesis-driven combination of LC-MS and NMR supported by computational validation tools.
Ready to advance your cyclic peptide characterization project? The analytical team at ResolveMass Laboratories Inc. offers more than two decades of practical experience in peptide structural confirmation, impurity profiling, and regulatory-grade analytical method development. Contact our scientists today to discuss a customized characterization strategy tailored to your cyclic peptide program.
Contact ResolveMass Laboratories Inc. →
Frequently Asked Questions (FAQs): Cyclic Peptide Characterization
Edman degradation relies on the presence of a free N-terminal amino group to initiate sequential cleavage and identification of amino acid residues. Since cyclic peptides possess a closed backbone structure, they do not contain an accessible N-terminus for the Edman reagent to react with. As a result, the chemistry required for stepwise sequencing cannot occur. To determine the sequence of cyclic peptides, researchers typically use advanced MSⁿ fragmentation strategies or NMR-based connectivity analysis.
Distinguishing cyclic peptides from their linear counterparts can be challenging because certain cyclic and linear forms may exhibit identical nominal masses. In these situations, fragmentation behavior becomes more informative than molecular weight alone. Energy-resolved MS/MS studies evaluate how each structure fragments under increasing collision energies, revealing characteristic differences between cyclic and linear molecules. Complementary analytical techniques, including mid-infrared spectroscopy, can provide additional confirmation when required.
Among all NMR techniques used for cyclic peptide analysis, HMBC is generally considered the most valuable for confirming ring connectivity. This experiment detects long-range proton-carbon correlations that extend across multiple chemical bonds, allowing researchers to observe interactions that span amide linkages. Because the ring-closing bond cannot be directly verified using COSY or HSQC experiments, HMBC provides the critical evidence needed to establish a closed cyclic structure. It is often the definitive experiment for validating successful cyclization.
D- and L-amino acids are mirror-image molecules that share identical molecular masses, making them indistinguishable by mass spectrometry alone. To determine stereochemistry, the cyclic peptide is first hydrolyzed into its constituent amino acids and then derivatized using a chiral reagent such as L-FDAA. This process converts the amino acids into diastereomeric derivatives that can be separated chromatographically. Comparison with authenticated standards enables accurate assignment of the configuration of each residue.
Single-stage MS² fragmentation often generates highly complex spectra because cyclic peptides can open at multiple positions within the ring. This produces overlapping fragment ion series that make sequence interpretation difficult. In contrast, MSⁿ allows a specific ring-opened fragment to be isolated and fragmented further in a controlled manner. By creating a directional sequence of fragmentation events, MSⁿ significantly improves sequence clarity and reduces ambiguity, especially for structurally complex cyclic peptides.
The quantity of material required depends on both the complexity of the peptide and the sensitivity of the NMR instrument. Basic proton-detected experiments such as COSY, TOCSY, and NOESY can often be performed with approximately 1–5 mg of purified sample. However, a more comprehensive characterization package that includes HMBC and carbon-based experiments generally benefits from 10–20 mg of material. For scarce natural products available only in trace amounts, LC-HRMS and MSⁿ frequently serve as the primary structural characterization tools.
Cyclic peptides often exhibit behavior that complicates chromatographic method development. Conformational changes can lead to broad or split peaks, while structurally similar cyclic and linear forms may co-elute due to comparable hydrophobicity. Highly hydrophobic peptides can also interact strongly with stationary phases, reducing chromatographic performance. Careful optimization of column chemistry, gradient conditions, mobile-phase composition, and operating temperature is typically required to achieve reliable separation and accurate purity assessment.
Cyclodepsipeptides contain ester linkages that behave differently from conventional amide bonds during analysis. In tandem mass spectrometry, ester bonds generally fragment more readily than amide bonds, generating characteristic product ions that help locate the linkage. NMR provides additional confirmation through the absence of an NH resonance at the ester position and the observation of diagnostic HMBC correlations. Together, these analytical signatures provide strong evidence for the presence and location of ester bonds within the molecule.
Reference:
- Stephanie, F., Tambunan, U.S.F., Kuczera, K., & Siahaan, T.J. (2024). Structure of a Cyclic Peptide as an Inhibitor of Mycobacterium tuberculosis Transcription: NMR and Molecular Dynamics Simulations. Pharmaceuticals, 17(11), 1545. https://doi.org/10.3390/ph17111545
- Maroto, A., Boqué, R., Jeanne Dit Fouque, D., & Memboeuf, A. (2024). Energy-Resolved Mass Spectrometry and Mid-Infrared Spectroscopy for Purity Assessment of a Synthetic Peptide Cyclised by Intramolecular Huisgen Click Chemistry. Methods and Protocols, 7(6), 97. https://doi.org/10.3390/mps7060097
- PLOS ONE (2012). mMass as a Software Tool for the Annotation of Cyclic Peptide Tandem Mass Spectra. https://doi.org/10.1371/journal.pone.0044913
- PMC (2016). Characterization of Cyclic Peptides Containing Disulfide Bonds by 2D NMR and ESI-MS/MS. PMC4608431. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4608431/
- Pevzner, P.A., et al. (2012). Sequencing Cyclic Peptides by Multistage Mass Spectrometry. PMC3398611. https://pmc.ncbi.nlm.nih.gov/articles/PMC3398611/
- Kalisz, O., et al. (2025). Origin and Characterization of Cyclodepsipeptides: Comprehensive Structural Approaches with Focus on Mass Spectrometry Analysis of Alkali-Cationized Molecular Species. PMC12502043. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12502043/

