
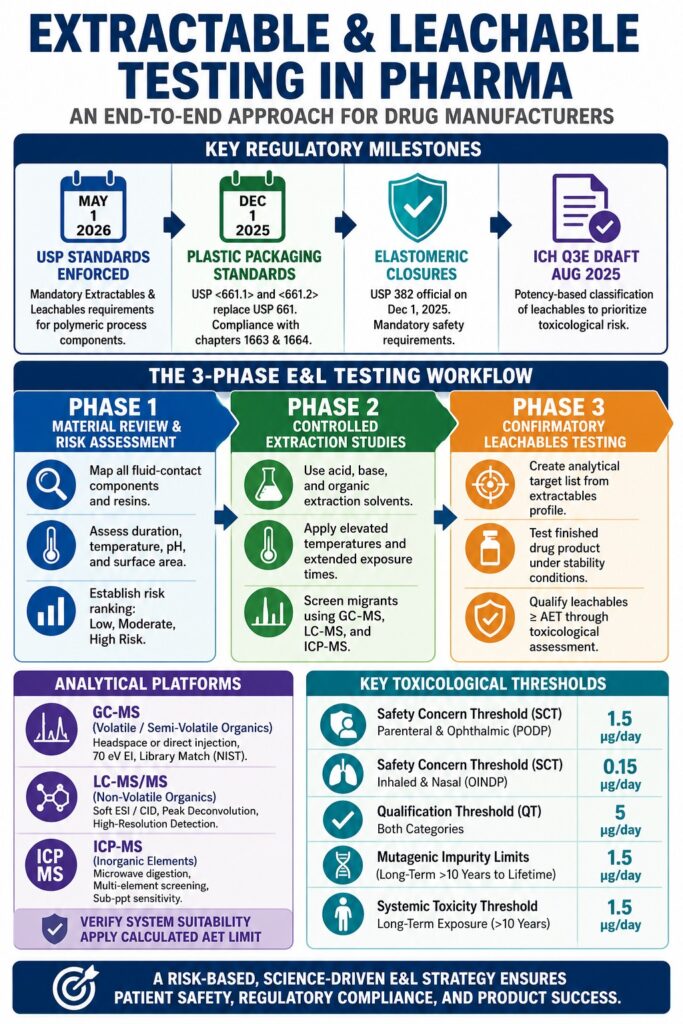
Regulatory Milestones Shaping Extractable and Leachable Testing in Pharma
The regulatory framework governing Extractable and Leachable Testing in Pharma has evolved from broad recommendations into highly standardized compendial requirements, primarily driven by the enforcement of USP standards on May 1, 2026. These standards require pharmaceutical manufacturers to systematically assess polymeric process components used during both drug substance and drug product manufacturing to control process-related impurities. Before this enforcement, expectations surrounding single-use technologies and process equipment were fragmented, compelling manufacturers to navigate numerous independent and largely voluntary guidance documents.
To better understand the core challenges and industry-standard approaches, learn more about the fundamentals of extractables and leachables in pharmaceutical products here.
Historically, global quality standards concentrated on evaluating final container closure systems, leaving a significant regulatory gap regarding chemical risks introduced during complex upstream and downstream bioprocessing operations. Under USP requirements and their accompanying explanatory chapters, manufacturers are now responsible for demonstrating that polymeric contact surfaces, including single-use bioreactor bags, filters, silicone tubing, connectors, and mixing assemblies, do not release Process Equipment-Related Leachables (PERLs) at concentrations that could compromise drug quality or patient safety.
This assessment requires a structured two-stage risk evaluation process. The first stage involves a contact assessment to identify all surfaces interacting with process fluids. The second stage includes a formal risk-ranking exercise based on factors such as contact duration, operating temperature, formulation pH, and the chemical composition of the process stream.
This regulatory transformation closely mirrors changes in plastic packaging evaluations. On December 1, 2025, the legacy USP 661 standard was officially replaced by USP <661.1>, which governs plastic materials of construction and resins, and USP <661.2>, which governs fully assembled plastic packaging systems. Compliance with USP <661.2> requires a comprehensive chemical safety assessment that incorporates the extractables and leachables principles outlined in USP chapters 1663 and 1664. At the same time, USP 382 became official on December 1, 2025, establishing mandatory functional and chemical safety requirements for elastomeric closures used in injectable drug products.
To bridge the gap between historical industry practices and current compendial requirements, many pharmaceutical companies compare the voluntary BioPhorum Operations Group (BPOG) protocol with the mandatory USP framework. Although BPOG has traditionally provided comprehensive extractables profiling, its highly prescriptive solvent systems and multiple time-point requirements often create substantial analytical and operational burdens compared with the risk-based methodology established under USP standards.
| Parameter | BPOG Protocol (2020 Update) | USP Standard |
|---|---|---|
| Regulatory Status | Voluntary industry best-practice framework | Mandatory United States Pharmacopeia compendial standard (May 1, 2026) |
| Scope of Materials | Polymeric and elastomeric single-use manufacturing components | Polymeric process components contacting drug streams |
| Extraction Solvents | Six solvents: Water, 5M NaCl, 0.1 M H₃PO₄, 0.5N NaOH, 50% ethanol, 1% Polysorbate 80 | Three model solvents: Acidified KCl/HCl (pH 3), Phosphate buffer (pH 10), 50% ethanol |
| Extraction Temperature | Standardized at 40°C and 121°C | Typically 40°C, adaptable to worst-case process temperatures |
| Time Points | Up to four intervals (e.g., 30 minutes, 24 hours, 21 days, 70 days) | Single scientifically justified worst-case exposure duration |
| Reporting Limits | Fixed surface-area normalized threshold (0.1 μg/cm²) | Analytical Evaluation Threshold (AET) scaled to clinical dosage |
The rationale behind these solvent selections illustrates the critical analytical considerations manufacturers must address. For example, comparisons between low-pH extraction profiles of polymeric filters demonstrate that BPOG’s 0.1 M H₃PO₄ and USP’s acidified KCl/HCl solution (pH 3) generate highly comparable extraction profiles for common organic extractables. However, because the KCl/HCl extraction matrix contains elevated concentrations of inorganic non-volatile salts, analytical laboratories typically implement a solvent delay period, often around one minute, during LC-MS analysis to divert corrosive salts away from the mass spectrometer source.
As a result, highly polar compounds that elute early, including triethylene glycol and the vulcanization stabilizer 4-hydroxy-2,2,6,6-tetramethyl-piperidine-1-ethanol (HTPE), may not be detected in acidified USP extracts. Alternative chromatographic strategies are therefore necessary to capture these highly polar migrants.
Similarly, comparisons between BPOG’s aggressive 0.5 N NaOH extraction system (pH approximately 13.5) and USP’s phosphate buffer (pH 10) reveal that the stronger alkaline environment is up to four times more efficient at extracting certain clarifying additives, including 1,3:2,4-bis(3,4-dimethylbenzylidene) sorbitol. Nevertheless, free phenol may remain undetected in 0.5 N NaOH because it exists predominantly in its ionized phenoxide form under strongly alkaline conditions, altering both its partitioning characteristics and chromatographic behavior.
In addition, BPOG’s inclusion of 1% Polysorbate 80 introduces substantial analytical challenges due to chromatographic co-elution and electrospray ionization (ESI) suppression during mass spectrometric analysis. By contrast, USP’s use of 50% ethanol provides a suitable organic extraction environment that correlates well with surfactant-containing formulations while avoiding the analytical complications associated with complex polyoxyethylene surfactants.
For a deeper dive into the specific compliance landscape, you can understand the requirements for USP extractables and leachables here.
Article Summary:
Article Summary (6–7 Key Points)
- Regulatory requirements for Extractables & Leachables (E&L) testing have become significantly stricter, with USP standards effective from May 2026 requiring pharmaceutical manufacturers to assess polymeric process-contact materials for potential chemical contamination risks.
- Modern E&L programs focus on a risk-based evaluation approach, requiring manufacturers to identify all product-contact surfaces and rank risks based on factors such as exposure time, temperature, pH, and process conditions.
- Three core analytical technologies support E&L studies: GC-MS for volatile and semi-volatile compounds, LC-MS/MS for non-volatile organic contaminants, and ICP-MS for trace-level elemental and inorganic impurities.
- The Analytical Evaluation Threshold (AET) is a critical decision-making tool, converting toxicological safety limits into analytical reporting thresholds to determine which chemical migrants require further investigation and qualification.
- Uncertainty factors are applied to AET calculations to account for variations in detector response and reduce the risk of overlooking potentially harmful compounds during screening studies.
- Toxicological risk assessments classify detected migrants according to their health hazards, helping manufacturers prioritize compounds that may present carcinogenic, mutagenic, reproductive, developmental, or systemic toxicity concerns.
- A complete E&L strategy follows a three-phase workflow: material risk assessment, controlled extractables studies under worst-case conditions, and confirmatory leachables testing on the finished drug product to ensure long-term safety, regulatory compliance, and product quality.
Analytical Methodologies Essential for Extractable and Leachable Testing in Pharma
Comprehensive Extractable and Leachable Testing in Pharma relies primarily on three analytical techniques: gas chromatography-mass spectrometry (GC-MS) for volatile compounds, liquid chromatography-mass spectrometry (LC-MS) for non-volatile compounds, and inductively coupled plasma-mass spectrometry (ICP-MS) for inorganic and elemental impurities. These complementary analytical platforms enable the detection of chemical migrants with widely varying molecular weights, polarities, and physicochemical properties at trace concentrations. Because extractables and leachables encompass diverse chemical classes, no single analytical technology can identify every potential contaminant.
To determine the most appropriate analytical approach for your specific drug product, compare GC-MS vs LC-MS in extractables and leachables testing here.
GC-MS serves as the primary analytical platform for volatile organic compounds (VOCs) and semi-volatile organic compounds (SVOCs). Volatile species, including residual polymerization solvents, monomer residues, and low-molecular-weight ink constituents, are generally analyzed using headspace GC-MS, which employs thermal desorption to release analytes directly from the polymer matrix. Semi-volatile compounds such as plasticizers, slip agents, and antioxidants are typically evaluated using direct liquid injection GC-MS.
The preferred ionization technique for GC-MS screening is electron ionization (EI) operating at a standardized energy of 70 eV. This high-energy process produces highly reproducible fragmentation patterns that can be compared against established spectral libraries such as the National Institute of Standards and Technology (NIST) database, enabling efficient compound identification.
For non-volatile and thermally sensitive organic compounds, including polymer oligomers, stabilizers, and degradation products, high-performance liquid chromatography coupled with high-resolution mass spectrometry (LC-MS) is required. Unlike EI-based GC-MS, electrospray ionization (ESI) used in LC-MS is a soft ionization technique that primarily generates protonated or deprotonated molecular ions.
To obtain meaningful structural information for compound identification, analysts perform LC-MS/MS experiments using multiple collision energies to induce collision-induced dissociation (CID). The resulting fragmentation spectra can vary significantly depending on instrument settings and collision energy conditions. Consequently, advanced peak deconvolution techniques are necessary to separate target analytes from background formulation signals.
Spectral deconvolution plays a critical role in trace-level pharmaceutical analysis because co-eluting excipients frequently suppress ionization or generate isobaric interferences.
[Polymeric Material Reflux / Digestion]
|
+------------------------+------------------------+
| | |
v v v
[Volatile Organics] [Non-Volatile Organics] [Inorganic Elements]
* Headspace GC-MS * Liquid LC-MS/MS * ICP-MS Digestion
* Standard 70 eV EI * Soft ESI / CID * Multi-Element Screening
* Library Match (NIST) * Peak Deconvolution * Sub-ppt Sensitivity
| | |
+------------------------+------------------------+
|
v
[Verify System Suitability]
[Apply Calculated AET Limit]Inorganic and elemental impurities, including catalyst residues, heavy metals, and glass-derived contaminants, are characterized using ICP-MS. According to USP chapters 232 and 233, ICP-MS remains the gold standard for elemental impurity analysis, providing detection limits in the sub-parts-per-trillion range, which are several orders of magnitude lower than those achievable with ICP-OES.
Sample preparation generally involves closed-vessel microwave digestion using concentrated nitric acid to completely dissolve complex polymeric and glass matrices into a homogeneous aqueous solution suitable for analysis.
To maintain analytical accuracy throughout these multidimensional workflows, the United States Pharmacopeia strongly emphasizes System Suitability Testing (SST) as part of E&L screening programs. Standardized reference materials and rubber oligomer Analytical Reference Materials (ARMs) developed by USP support system suitability verification and compound identification. These materials ensure consistent instrument sensitivity, chromatographic performance, and mass accuracy throughout analytical studies.
Calculating the Analytical Evaluation Threshold in Extractable and Leachable Testing in Pharma
The Analytical Evaluation Threshold (AET) is determined by converting a dose-based toxicological threshold, such as the Safety Concern Threshold (SCT), into a concentration-based reporting limit using the maximum daily dose and an analytical uncertainty factor. This approach ensures that only compounds with genuine toxicological relevance are identified and evaluated, minimizing both false negatives and unnecessary analytical complexity.
The AET serves as the concentration boundary below which a chemical migrant is considered to present negligible toxicological concern, eliminating the need for additional chemical characterization or toxicological assessment.
To establish the initial unadjusted AET (AET_initial) in units of μg/mL, the following equation is applied:
AET_initial = (SCT / MDD) × (V_extract / (V_contact × f))
Where:
- SCT represents the Safety Concern Threshold.
- MDD represents the Maximum Daily Dose.
- V_extract is the extraction solvent volume.
- V_contact is the total contact volume or surface area of the component.
- f is the accumulation factor accounting for multiple contributing components.
An alternative estimation approach based on container closure system parameters is:
AET_estimated = (SCT / Doses_day) × (Doses_CCS / Weight_CCS)
Where:
- Doses_day is the number of daily doses administered.
- Doses_CCS is the total number of doses within a single container closure system.
- Weight_CCS represents the mass, volume, or surface area of the contact component.
Selection of the Safety Concern Threshold depends heavily on the route of administration and duration of exposure. For Parenteral and Ophthalmic Drug Products (PODP), PQRI recommends a standard SCT of 1.5 μg/day. For Orally Inhaled and Nasal Drug Products (OINDP), a significantly lower SCT of 0.15 μg/day is recommended due to the heightened sensitivity of pulmonary tissues and direct systemic exposure.
To guide your team through the necessary variables and formulas, learn how to calculate the AET for your studies here.
The Qualification Threshold (QT) is generally established at 5 μg/day for both categories and represents the exposure level below which non-mutagenic leachables typically do not require comprehensive toxicological qualification.
FDA draft guidance for topical ophthalmic solutions recommends the following concentration-based thresholds:
| Threshold Type | Recommended Limit |
| Reporting Threshold | 1 ppm |
| Identification Threshold | 10 ppm |
| Qualification Threshold | 20 ppm |
When evaluating potential mutagenic impurities under ICH M7 and draft ICH Q3E guidance, acceptable exposure limits decrease as treatment duration increases. This risk-based approach helps control cumulative mutagenic risk throughout a patient’s lifetime.
| Clinical Treatment Duration | Acceptable Daily Intake (Single Mutagenic Impurity) | Acceptable Total Daily Intake (Multiple Mutagenic Impurities) |
| ≤ 1 Month | 120 μg/day | 120 μg/day |
| >1 to 12 Months | 20 μg/day | 60 μg/day |
| >1 to 10 Years | 10 μg/day | 30 μg/day |
| >10 Years to Lifetime | 1.5 μg/day | 5 μg/day |
Systemic toxicity thresholds also vary according to treatment duration. For long-term exposure exceeding ten years, the non-cancer systemic toxicity threshold is 1.5 μg/day for oral, parenteral, dermal, and inhalation routes. This limit increases to 10 μg/day for treatment periods greater than one year but less than ten years, and to 20 μg/day for treatment durations between one month and one year.
Applying the Analytical Uncertainty Factor
E&L screening methods often rely on relative quantitation using a single internal reference standard. This approach assumes that both the reference standard and unknown migrants produce equivalent detector responses. In practice, however, chemical compounds exhibit widely varying Relative Response Factors (RRFs).
If a hazardous compound has a substantially lower RRF than the reference standard, its actual concentration may exceed safety thresholds even when its measured peak area falls below the calculated limit. This scenario creates the potential for a Type II false-negative result.
To address this risk, the calculated AET is adjusted downward using an Uncertainty Factor (UF):
AET_adjusted = AET_initial / UF
The Uncertainty Factor may be derived from the Relative Standard Deviation (RSD) of response factors within a validated compound database:
UF = 1 / (1 − RSD)
Historically, a default UF value of 2, representing a 50% safety margin, was commonly accepted for routine pharmaceutical analyses, particularly in GC-FID and GC-MS applications where response factors are relatively consistent. However, modern regulatory frameworks, including ISO 10993-18 and draft ICH Q3E guidance, discourage the use of fixed default values in favor of experimentally justified, method-specific uncertainty factors.
For highly variable analytical methods such as electrospray ionization LC-MS, RSD values can be considerably higher, requiring uncertainty factors ranging from 5 to 10. If the calculated UF exceeds 10, it may indicate that the analytical method lacks adequate robustness and could reduce the adjusted AET below the instrument’s limit of detection, necessitating method refinement or targeted quantitative validation.
Toxicological Risk Assessment of Chemical Migrants in Extractable and Leachable Testing in Pharma
Toxicological risk assessment within Extractable and Leachable Testing in Pharma evaluates the biological safety of identified migrants by comparing estimated patient exposure levels against established thresholds such as the Safety Concern Threshold (SCT) and Permitted Daily Exposure (PDE). This process categorizes compounds according to hazard potential and helps identify carcinogenic, mutagenic, reproductive, developmental, and immunogenic risks.
The draft ICH Q3E guideline, released in August 2025, introduces a potency-based classification framework for organic leachables. This approach enables pharmaceutical manufacturers to prioritize impurities according to toxicological risk.
| Toxicological Class | Risk Profile | Examples | Required Action |
| Class 1 (Avoid) | Potent mutagenic carcinogens, DNA-reactive agents, or highly potent toxicants | NDBA, NDMA, Benzo[a]pyrene | Avoid whenever possible and maintain exposure below compound-specific acceptable intake limits |
| Class 2 (Limit) | Reproductive, developmental, systemic, organ-specific toxicants, or irritants | DEHP, BPA, MBT | Establish compound-specific PDE values |
| Class 3 (Low) | Minimal toxicological concern | MOSH, synthetic polymer oligomers | Standard toxicological qualification |
Chemical migration can produce two primary categories of clinical risk. The first is direct toxicity, in which the migrant itself possesses inherent toxicological hazards. The second is indirect or product-mediated toxicity, in which the migrant interacts chemically with the drug product, leading to degradation of the active pharmaceutical ingredient or formation of immunogenic complexes.
Phthalate plasticizers such as bis(2-ethylhexyl) phthalate (DEHP), commonly used to enhance flexibility in polyvinyl chloride (PVC) materials, are well-recognized endocrine-disrupting compounds associated with developmental and reproductive toxicity. Similarly, Bisphenol A (BPA), frequently found in polycarbonate plastics and epoxy resins, exhibits endocrine activity through interactions with estrogen receptors.
Given the complexity of these assessments, ensure safety and regulatory alignment with professional toxicological qualification of leachables here.
Among the most concerning organic migrants are N-nitrosamines such as N-nitrosodibutylamine (NDBA) and N-nitrosodimethylamine (NDMA), both of which are classified as Class 1 compounds under ICH Q3E due to their potent mutagenic and carcinogenic properties. These contaminants often originate from vulcanization accelerators used during the manufacture of elastomeric closures and sealing materials.
Polycyclic aromatic hydrocarbons (PAHs), including benzo[a]pyrene, may also migrate from carbon black pigments used in polymeric materials.
The significance of product-mediated degradation has been demonstrated in historical cases involving organic sulfur compounds, particularly vulcanization agents such as dialkylphenol disulfide, that leached from uncoated rubber syringe plungers. These compounds reacted with the drug formulation, producing adjuvant-like effects that reduced therapeutic efficacy and contributed to severe immunogenic responses, including pure red cell aplasia.
Designing an End-to-End Protocol for Extractable and Leachable Testing in Pharma
A comprehensive E&L testing strategy begins with a detailed material risk assessment, progresses through controlled extractables profiling under worst-case laboratory conditions, and concludes with stability-indicating confirmatory leachables testing. This structured workflow supports regulatory compliance while minimizing the likelihood of late-stage manufacturing failures, regulatory setbacks, or clinical delays.
[Phase 1: Material Review & Risk Assessment]
- Map all fluid-contact components and resins
- Assess duration, temperature, pH, and surface area
- Establish risk ranking (Low, Moderate, High Risk)
|
v
[Phase 2: Controlled Extraction Studies]
- Select acid, base, and organic extraction solvents
- Apply elevated temperatures and extended exposure times
- Screen migrants using GC-MS, LC-MS, and ICP-MS
|
v
[Phase 3: Confirmatory Leachables Testing]
- Create analytical target list from extractables profile
- Test finished drug product under stability conditions
- Qualify leachables exceeding the AET through toxicological assessmentPlanning for these stages requires significant resource allocation; you can get an expert guide on the cost of extractables and leachables testing to plan your budget here.
Phase 1: Material Review and Risk Assessment
The first phase involves identifying and documenting all fluid-contact surfaces throughout the manufacturing process or packaging system, including polymer resins, additives, inks, coatings, and adhesives. Each component is assigned a risk ranking based on process location, proximity to the final dosage form, operating conditions, contact duration, and the leaching potential of the formulation.
Low-risk components, such as polymeric systems used in oral liquid manufacturing that comply with applicable food-contact regulations, may often be qualified through abbreviated data packages or compendial compliance alone. High-risk components, including single-use bioreactor bags, sterile process filters, and primary container closures used with parenteral products, require extensive chemical characterization.
Phase 2: Controlled Extraction Studies
Controlled extraction studies expose polymeric components to exaggerated laboratory conditions using multiple extraction solvents, elevated temperatures, and extended exposure durations. The objective is to establish a comprehensive chemical profile of all potential migrants and generate an Analytical Target List (ATL) for confirmatory leachables testing.
Typical extractables studies require approximately 8 to 12 weeks to complete, encompassing method development, extraction execution, data processing, and chromatographic deconvolution. To support compliance with compendial standards, pharmaceutical manufacturers often collaborate with specialized analytical laboratories such as ResolveMass Laboratories Inc. to design scientifically justified extraction protocols and verify analytical performance requirements.
Phase 3: Confirmatory Leachables Testing
Confirmatory leachables studies evaluate the actual migration of chemical species into the finished drug product under commercial storage, transportation, usage, and stability conditions. These studies are generally conducted alongside real-time stability programs and may extend from 3 to 12 months or throughout the product’s intended shelf life using validated stability-indicating analytical methods.
Any detected leachable that meets or exceeds the calculated AET must be identified, quantified, and subjected to formal toxicological qualification. Ideally, the observed leachables profile represents a subset of the extractables profile generated during earlier studies. However, additional leachables may emerge over time due to formulation interactions, material aging, sterilization effects, or degradation processes, highlighting the importance of long-term monitoring throughout product development and commercialization.
Strategic Execution and Compliance for Extractable and Leachable Testing in Pharma
Successful execution of Extractable and Leachable Testing in Pharma depends on integrating a comprehensive risk-based study design with advanced high-resolution analytical techniques capable of detecting trace-level organic and inorganic compounds. Establishing a scientifically sound and well-documented compliance strategy is essential for minimizing the risk of regulatory setbacks, product batch rejections, and potential patient safety issues. Pharmaceutical manufacturers should consider chemical compatibility assessments a critical component of their Chemistry, Manufacturing, and Controls (CMC) strategy, ensuring that material-related risks are addressed throughout the product development lifecycle.
By adopting a comprehensive end-to-end testing program aligned with USP standards and draft ICH Q3E guidance, manufacturers can develop robust baseline extractables profiles that support supplier qualification efforts, strengthen raw material control strategies, and facilitate efficient regulatory submissions across global markets. This proactive and science-driven approach allows organizations to identify and resolve potential challenges, including material incompatibilities, additive migration, and formulation degradation, during the early stages of development. Addressing these risks proactively helps prevent costly clinical delays, manufacturing disruptions, and post-commercialization product issues.
ResolveMass Laboratories Inc. utilizes advanced chromatographic and spectrometric technologies to support complex analytical activities, including peak deconvolution, Analytical Evaluation Threshold (AET) calculations, and comprehensive toxicological assessments. Collaborating with a specialized Extractables and Leachables testing partner helps ensure that study designs are scientifically justified, compliant with applicable compendial requirements, and appropriately tailored to the clinical dosing characteristics and risk profile of each therapeutic product.
For organizations seeking support in developing USP or ICH Q3E-compliant Extractables and Leachables testing programs, additional guidance and consultation can be obtained through the ResolveMass Contact Us page.
Frequently Asked Questions Regarding Extractable and Leachable Testing in Pharma
USP becomes an enforceable requirement on May 1, 2026. The standard applies to pharmaceutical manufacturers and biopharmaceutical companies that use polymeric process equipment, disposable manufacturing assemblies, or single-use technologies during drug substance or drug product production. Organizations utilizing fluid-contact polymeric components must demonstrate compliance with the new requirements. The regulation is intended to strengthen control over process-related chemical impurities and improve patient safety.
USP <661.1> focuses on evaluating plastic materials at the raw material stage, including the resin and material composition before the manufacturing process. In contrast, USP <661.2> applies to the finished plastic packaging system after assembly and fabrication. Regulatory compliance is generally assessed at the packaging-system level under USP <661.2>. Even when individual materials do not fully conform to USP <661.1>, the final system may still be acceptable if it demonstrates adequate chemical and biological safety.
The BPOG protocol incorporates 1% Polysorbate 80 to mimic the extraction behavior of highly surfactant-rich and lipophilic biopharmaceutical formulations. USP utilizes 50% ethanol because it provides an effective organic extraction environment while maintaining analytical simplicity. Polysorbate 80 can create challenges during mass spectrometric analysis due to ion suppression and overlapping chromatographic peaks. As a result, 50% ethanol often provides cleaner analytical data while producing comparable extractables information.
The Safety Concern Threshold (SCT) represents an exposure level below which a leachable is considered to present minimal toxicological concern, regardless of its chemical identity. Typical SCT values are 1.5 μg/day for parenteral products and 0.15 μg/day for orally inhaled and nasal drug products. The Qualification Threshold (QT), generally set at 5 μg/day, defines the level below which a non-mutagenic leachable usually does not require extensive toxicological qualification. Together, these thresholds help determine the level of investigation required for detected compounds.
ICH M7 applies a risk-based approach in which acceptable exposure limits for mutagenic impurities become more restrictive as treatment duration increases. For short-term therapies lasting one month or less, a higher daily intake level is considered acceptable. However, for long-term or lifetime treatment scenarios, the permissible daily exposure is substantially reduced. This framework is designed to minimize cumulative mutagenic risk over a patient’s lifetime while maintaining an appropriate balance between safety and therapeutic benefit.
Secondary leachables are compounds that develop after manufacturing as a result of chemical interactions between primary leachables and components of the drug formulation. These substances may not be present during initial extractables studies and can emerge gradually throughout product storage. Their formation can be influenced by excipients, active pharmaceutical ingredients, or environmental conditions. Detecting secondary leachables generally requires stability-indicating analytical methods capable of monitoring changes in the formulation over time.
The FDA draft guidance for topical ophthalmic drug products establishes concentration-based limits specifically designed for this sensitive route of administration. A reporting threshold of 1 ppm is recommended for documenting detected compounds. An identification threshold of 10 ppm is applied when compound characterization becomes necessary, while a qualification threshold of 20 ppm is used to determine when toxicological assessment is required. These limits help ensure the safety and quality of ophthalmic formulations.
A solvent delay is commonly incorporated during LC-MS analysis to prevent concentrated potassium and chloride salts from entering the mass spectrometer source. Diverting these inorganic salts to waste helps protect the instrument and reduces contamination-related maintenance issues. However, the delay period can also result in the loss of analytical information for compounds that elute very early during chromatography. Consequently, certain highly polar extractables may require alternative analytical approaches to ensure detection.
Reference:
- Kuzmič, S., Zlobec, T., Sollner Dolenc, M., Roškar, R., & Trdan Lušin, T. (2026). Extractables and leachables in pharmaceutical products: Potential adverse effects and toxicological risk assessment. Toxics, 14(1), 92. https://doi.org/10.3390/toxics14010092
- United States Pharmacopeia. (n.d.). Extractables and leachables. USP. https://www.usp.org/impurities/extractables-and-leachables
- European Medicines Agency. (2026). Overview of comments received on ICH Q3E guideline and supporting documentation for extractables and leachables (EMA/CHMP/ICH/236669/2025 and EMA/CHMP/ICH/236668/2025) (EMA/2613/2026 Rev. 1). European Medicines Agency. https://www.ema.europa.eu/en/documents/comments/overview-comments-received-ich-q3e-guideline-supporting-documentation-extractables-leachables-ema-chmp-ich-236669-2025-ema-chmp-ich-236668-2025_en.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2025, September). ICH Q3E: Guideline for extractables and leachables—Step 2 draft guideline (presentation). https://database.ich.org/sites/default/files/ICH_Q3E_Step2_Presentation_2025_0826.pdf

