
Understanding the Regulatory Landscape of FDA Questions on E&L Studies
The Food and Drug Administration (FDA) carefully evaluates extractables and leachables (E&L) data to ensure that chemical substances migrating from packaging materials, manufacturing equipment, or drug delivery systems do not adversely affect the safety, quality, or efficacy of pharmaceutical products. To meet these expectations, pharmaceutical organizations develop E&L programs that align with evolving regulatory standards, including USP <1663>, USP <1664>, and the draft ICH Q3E guideline.
Within today’s highly regulated pharmaceutical environment, preparing for potential FDA Questions on E&L Studies has evolved from a packaging-related activity conducted near the end of development into a critical regulatory requirement managed throughout the product lifecycle. Chemical migration from container-closure systems (CCS), drug delivery devices, and single-use manufacturing components can present significant risks to both patient safety and product performance. As a result, the FDA reviews E&L data with increasing scrutiny and scientific rigor.
To reduce the likelihood of receiving Complete Response Letters (CRLs), clinical holds, or regulatory approval delays, sponsors must establish scientifically sound justifications, validated analytical methodologies, and well-supported toxicological safety thresholds. A proactive and comprehensive E&L strategy is now considered an essential element of successful pharmaceutical development and regulatory submission.
Learn how to design compliant testing frameworks by reading our definitive guide on Extractables and Leachables (E&L) Testing for Drug Safety for NDA/ANDA Submissions.
Article Summary:
- The FDA closely reviews Extractables and Leachables (E&L) data to ensure that chemicals migrating from packaging, manufacturing systems, or drug delivery devices do not compromise product safety, quality, or effectiveness.
- Modern pharmaceutical companies must treat E&L assessment as a lifecycle-wide regulatory requirement rather than a late-stage packaging activity, as inadequate E&L programs can lead to approval delays, Complete Response Letters (CRLs), or clinical holds.
- Key regulatory frameworks such as USP <1663>, USP <1664>, ISO 10993-18, and the draft ICH Q3E guideline provide the scientific and regulatory foundation for extractables testing, leachables studies, and toxicological risk evaluation.
- A proactive E&L strategy typically includes material risk assessment, controlled extractables studies, Analytical Evaluation Threshold (AET) determination, analytical method validation, and long-term leachables monitoring during product stability studies.
- Different packaging and manufacturing materials present unique migration risks. Common concerns include plasticizers, antioxidants, degradation products, metals, nitrosamines, and other processing-related compounds that may enter the drug product.
- FDA reviewers expect extractables studies to use scientifically justified worst-case extraction conditions, including multiple solvents, elevated temperatures, and extended exposure periods to maximize the detection of potential chemical migrants.
- Accurate AET calculation and analytical method validation are essential. Testing methods must demonstrate sufficient sensitivity to detect trace-level compounds below toxicological reporting thresholds while accounting for analytical variability through justified uncertainty factors.
- When FDA deficiencies arise, sponsors should provide data-driven responses supported by compound identification, toxicological assessments, Margin of Safety (MOS) calculations, validated analytical methods, and additional testing where necessary to demonstrate patient safety and regulatory compliance.
Standard E&L Regulatory Frameworks and Industry Standards
Successfully navigating the E&L regulatory environment requires a thorough understanding of the key pharmacopeial and international standards that govern extractables testing, leachables assessments, and toxicological evaluations. These frameworks establish the scientific foundation for identifying potential chemical migrants and determining whether patient exposure remains within acceptable safety limits.
| Regulatory Guideline | Target Scope | Core Methodological Expectations | Impact on FDA Submissions |
|---|---|---|---|
| USP <1663> Extractables Assessment | Establishes scientific principles for controlled laboratory extraction studies. | Defines the worst-case chemical profile required to identify potential migrants. | Provides the foundational extractables dataset expected during regulatory review. |
| USP <1664> Leachables Assessment | Requires real-time or simulated-use testing of drug products throughout shelf-life stability studies. | Verifies actual patient exposure levels under labeled storage conditions. | Demonstrates the real-world impact of chemical migration on the final drug product. |
| ISO 10993-18 Medical Device Characterization | Requires chemical characterization of patient-contacting materials based on exposure duration and use conditions. | Supports identification and assessment of material-related chemical risks. | Directly applicable to drug-delivery devices, prefilled syringes, and combination products. |
| ICH Q3E (Draft) Global E&L Harmonization | Introduces a lifecycle-based risk management framework aligned with ICH Q9 principles. | Emphasizes proactive risk assessment, material characterization, and exposure-based control strategies. | Harmonizes global regulatory expectations and promotes consistent E&L practices across regions. |
Historically, E&L assessments were often viewed as retrospective packaging qualification activities performed near the conclusion of product development. That approach is no longer sufficient. A significant portion of FDA Complete Response Letters now reference deficiencies associated with E&L programs, indicating that reactive management of chemical migration risks is increasingly unacceptable from a regulatory perspective.
Understand the baseline pharmacopeial requirements for submission readiness by exploring USP Extractables and Leachables Guidelines.
The current regulatory expectation emphasizes a proactive, science-driven approach that integrates material characterization, highly sensitive analytical testing, and comprehensive toxicological risk assessment from the earliest stages of product development. Organizations that adopt this strategy are better positioned to address regulatory concerns before they affect development timelines.
Ensure your regulatory documentation meets geography-specific frameworks by reviewing our brief on E&L Testing in the United States.
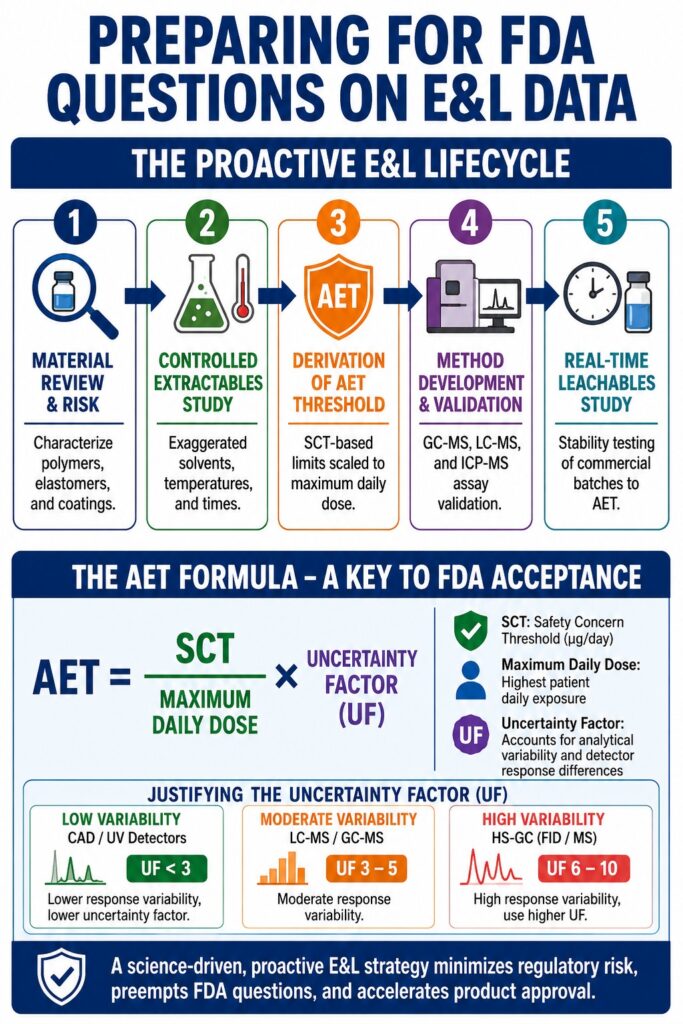
Proactive Strategies for Addressing FDA Questions on E&L Studies
Organizations can significantly reduce regulatory risk by conducting controlled extractables studies under exaggerated extraction conditions to establish a comprehensive chemical profile of potential migrants. These studies generate baseline data that support the design of focused leachables programs and real-time stability studies that monitor actual chemical migration throughout the product shelf life.
By identifying and characterizing potential risks before filing an Investigational New Drug (IND) application or New Drug Application (NDA), sponsors can proactively address concerns that might otherwise result in regulatory deficiencies.
┌────────────────────────────────────────────────────────┐
│ 1. Material Review & Risk │
│ (Characterize polymers, elastomers, and coatings) │
└───────────────────────────┬───────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────┐
│ 2. Controlled Extractables Study │
│ (Exaggerated solvents, temperatures, and times) │
└───────────────────────────┬───────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────┐
│ 3. Derivation of AET Threshold │
│ (SCT-based limits scaled to maximum daily dose) │
└───────────────────────────┬───────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────┐
│ 4. Method Development & Validation │
│ (GC-MS, LC-MS, and ICP-MS assay validation) │
└───────────────────────────┬───────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────┐
│ 5. Real-Time Leachables Study │
│ (Stability testing of commercial batches to AET) │
└────────────────────────────────────────────────────────┘
Discover how proactive planning ensures market access by checking out our overview on Extractables and Leachables (E&L) Requirements for U.S. Market Authorization.
Material-Specific E&L Risks and Target Analytes
Different polymeric and elastomeric materials used in pharmaceutical manufacturing and packaging exhibit unique chemical migration profiles. Understanding the composition and processing history of these materials enables developers to focus on compounds most likely to migrate into the drug product.
| Contact Material Category | Typical Pharmaceutical Application | Inherent Chemical Risks / Target Analytes | Essential Analytical Technique |
|---|---|---|---|
| Elastomeric Closures | Stopper systems, syringe plungers, vial caps | Vulcanizing agents, sulfur-containing compounds, zinc, plasticizers, n-nitrosamines | GC-MS and ICP-MS |
| Polymer Bags & Tubing | Single-use bioprocessing bags, manufacturing lines | Antioxidants, Irganox degradation products, slip agents, processing aids | LC-MS/MS and HS-GC/MS |
| Low-Density Polyethylene (LDPE) | Ophthalmic squeeze bottles, inhalation containers | Volatile migrants such as vanillin and adhesive-related compounds | Headspace GC-MS |
| Polyvinyl Chloride (PVC) | Infusion lines, IV bags, parenteral administration systems | Plasticizer migration, particularly phthalates such as DEHP | LC-MS/MS and FTIR |
Learn how material selection impacts modern drug modalities by exploring Extractables and Leachables in Biologics and ATMPs.
Worst-Case Extraction Parameter Selection
The design of an extractables study requires careful selection of extraction solvents, temperatures, and durations that represent worst-case exposure conditions relative to the intended clinical use of the drug product. FDA reviewers frequently challenge studies that utilize weak extraction conditions or insufficiently justified solvent systems.
For example, when evaluating a water-based injectable product, extractables testing should include solvents spanning a broad range of polarities, such as water, ethanol, and hexane. This approach improves the likelihood of detecting polar, semi-volatile, and non-volatile compounds that may migrate from contact materials.
Extraction temperatures should simulate accelerated thermodynamic conditions, including autoclaving, refluxing, or prolonged exposure to temperatures between 50°C and 60°C. These conditions are intended to mimic long-term interactions between the formulation and packaging materials.
When extraction conditions begin to compromise the structural integrity of a material, sponsors must clearly document how parameters were optimized to maximize chemical recovery while avoiding excessive polymer degradation. A well-supported scientific rationale is essential for regulatory acceptance.
Analytical Method Validation to Preempt FDA Questions on E&L Studies
Comprehensive analytical method validation is essential for demonstrating that testing methodologies possess the required sensitivity, precision, accuracy, and specificity to detect trace-level impurities. A critical aspect of validation is proving that the Limit of Quantitation (LOQ) remains sufficiently below the calculated Analytical Evaluation Threshold (AET).
If method validation fails to establish that the analytical platform can reliably detect compounds at concentrations relevant to patient safety, the FDA may issue significant deficiencies during NDA or ANDA review. Consequently, validation protocols must explicitly demonstrate the relationship between instrument performance and the toxicological thresholds used within the E&L program.
Mathematical Derivation of the Analytical Evaluation Threshold
The calculation and scientific justification of the Analytical Evaluation Threshold (AET) represent one of the most important elements of any E&L program. The AET establishes the reporting threshold for unknown chemical peaks and is directly influenced by the Safety Concern Threshold (SCT), patient dosing regimen, and analytical uncertainty.
For parenteral products, the SCT is typically established at 1.5 μg/day, while higher-risk ophthalmic and inhalation products commonly utilize a threshold of 0.15 μg/day.
The primary equation used to calculate the AET is:
[
\text{AET} = \frac{\text{SCT}}{\text{Maximum Daily Dose}} \times \text{Uncertainty Factor}
]
For more complex delivery systems, including prefilled syringes and nasal spray devices, the calculation is often expanded to account for component weight or volume:
[
\text{AET}\left(\frac{\mu g}{g}\right) =
\left(
\frac{\text{SCT}}{\text{Doses per Day}}
\times
\frac{\text{Doses per CCS}}{\text{Weight of CCS}}
\right)
\times
\text{Uncertainty Factor}
]
Where:
- SCT represents the Safety Concern Threshold expressed in μg/day.
- Doses per Day refers to the maximum anticipated daily patient exposure.
- Doses per CCS represents the number of doses associated with the container-closure system.
- Weight of CCS corresponds to the physical mass of the packaging component contacting the formulation.
- Uncertainty Factor (UF) is applied to compensate for analytical variability and differences in detector response among chemical species.
Justifying the Uncertainty Factor and Addressing Detection Bias
One of the most common triggers for FDA Questions on E&L Studies involves inadequate justification of the Uncertainty Factor used in AET calculations. Because E&L screening often relies on semi-quantitative analysis using a limited set of reference standards, detector responses can vary substantially among different compounds.
These variations are commonly expressed as Relative Response Factors (RRFs). When a compound exhibits a low response factor relative to the selected internal standard, its true concentration may exceed toxicological safety limits while still producing an analytical signal below the standard AET. This creates the risk of false-negative results and potential underestimation of patient exposure.
Signal Intensity (Abundance)
▲
│ ┌───┐ True Concentration (Above safety limit)
│ │ │
│ │ │ ◄─── Low response factor causes signal
│ └───┘ to fall below standard AET.
│ ───────────────────────────┬─────────────────────────── Standard AET
│ ▼
│ ┌───┐ Measured Signal (Misdetected as safe)
│ └───┘
│ ───────────────────────────┬─────────────────────────── Adjusted AET (with UF)
│ │ ◄─── Applying Uncertainty Factor (UF)
│ ▼ lowers threshold, ensuring detection.
└──────────────────────────────┴───────────────────────────► Time (Retention)
To minimize detection bias, the AET should be adjusted using a scientifically justified UF. ISO 10993-18:2020 recommends deriving this factor through statistical evaluation of the laboratory’s internal RRF database, ensuring that it reflects actual analytical performance.
FDA research has shown that Charged Aerosol Detection (CAD) and ultraviolet (UV) chromatography systems generally exhibit lower response variability and may require a UF below 3. In contrast, Headspace Gas Chromatography (HS-GC/FID or HS-GC/MS) often demonstrates greater variability, resulting in recommended UFs between 6 and 10.
Electrospray Ionization Mass Spectrometry (ESI-MS) presents additional challenges because ionization efficiency can vary dramatically among compounds. As a result, FDA reviewers frequently reject simplistic UF-based approaches when ESI-MS serves as the primary detection platform. In these situations, sponsors should consider alternative strategies such as multi-detector analytical systems, corrected RRF models, or compound-specific assays to ensure reliable detection of potentially harmful migrants.
Understand how to manage specialized impurities like nitrosamines stemming from packaging materials by checking out Packaging Leachables and Nitrosamine E&L Mitigation.
Formulating Robust Answers to Common FDA Questions on E&L Studies
Successfully resolving FDA deficiencies requires a structured response strategy that links each reviewer concern to a scientifically justified analytical or toxicological explanation. Responses should be supported by objective evidence, including corrected response factors, method validation data, exposure assessments, and Margin of Safety (MOS) calculations.
When a deficiency letter is received, sponsors should systematically evaluate each question, identify underlying data gaps, and implement targeted corrective actions designed to address the specific regulatory concern.
Standard Deficiency Response Workflow
A structured workflow enables development teams to manage E&L-related deficiencies efficiently while minimizing impacts on approval timelines.
┌────────────────────────────────────────────────────────┐
│ 1. Map Deficiencies to Working Matrix │
│ (Identify questions, severity, and analytical gaps) │
└───────────────────────────┬────────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────┐
│ 2. Evaluate Feasibility of Data Bridging │
│ (Justify material equivalency or historical data use) │
└───────────────────────────┬────────────────────────────┘
│
┌─────────────────┴─────────────────┐
▼ ▼
(Bridging Feasible) (Bridging Not Feasible)
┌───────────────────────────┐ ┌───────────────────────────┐
│ Compile Scientific Support│ │ Perform Targeted Testing │
│ • Supplier declarations │ │ • Extended extractions │
│ • Process equivalency │ │ • Multi-solvent screening │
│ • Material database refs │ │ • Method LOQ validation │
└─────────┬─────────────────┘ └─────────┬─────────────────┘
│ │
└─────────────────┬────────────────┘
│
▼
┌────────────────────────────────────────────────────────┐
│ 3. Execute Toxicological Risk Assessment │
│ (Derive PDEs and MOS calculations) │
└───────────────────────────┬────────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────┐
│ 4. Compile and Submit Response Book │
│ (Technical response package with supporting annexes) │
└────────────────────────────────────────────────────────┘
Addressing Unknown Peaks Above the AET
A frequently encountered FDA deficiency states:
“Several unidentified chromatographic peaks were observed above the calculated AET in the stability studies of the drug product. Identify these compounds and provide a toxicological risk assessment.”
Sponsors should avoid simply resubmitting previously generated data. Instead, the issue should be addressed through a structured identification strategy that includes:
High-Resolution Mass Spectrometry (HRMS)
Employing Orbitrap or Quadrupole Time-of-Flight (Q-TOF) mass spectrometry to obtain accurate mass measurements and isotopic distributions, enabling determination of molecular formulas.
Spectral Database Matching
Comparing acquired fragmentation spectra against established databases such as NIST or ELSIE to identify potential chemical structures.
Confirmation Using Reference Standards
Obtaining or synthesizing authentic reference materials and confirming identity through retention-time matching and fragmentation-pattern comparison.
Calculating and Justifying the Margin of Safety
When a leachable compound is detected above the AET, its toxicological significance must be evaluated through calculation of the Margin of Safety (MOS). This assessment considers the route of administration, duration of exposure, and maximum patient exposure levels.
The MOS is calculated using the following equation:
[
\text{MOS} =
\frac{\text{PDE}\left(\mu g/day\right)}
{\text{Maximum Daily Patient Exposure}\left(\mu g/day\right)}
]
Where:
- PDE (Permitted Daily Exposure) is derived from toxicological studies using NOAEL or LOAEL data and adjusted with appropriate uncertainty factors according to ICH Q3D and ICH Q3E principles.
- Maximum Daily Patient Exposure represents the highest estimated patient exposure based on measured leachable concentrations at the end of shelf life and the maximum approved daily dose.
An MOS greater than 1 generally indicates that exposure levels are toxicologically acceptable and present negligible risk to patients. Conversely, an MOS below 1 may require additional toxicological justification, material modifications, manufacturing process changes, or implementation of a dedicated control strategy.
To learn more about assessing structural alerts and downstream safety thresholds, review our specialized analysis on Extractables and Leachables Carcinogenicity Testing.
Resolving E&L Deficiencies with ResolveMass Laboratories Inc.
When pharmaceutical developers encounter complex FDA Questions on E&L Studies, collaboration with a specialized contract research organization can provide the expertise necessary to generate regulatory-ready solutions. ResolveMass Laboratories Inc., a USFDA-registered and ISO 9001:2015-certified facility located in Laval, Canada, offers comprehensive support for pharmaceutical development and advanced E&L investigations.
Examine real-world enforcement trends and responses by checking out our deep dive into FDA Extractables and Leachables Case Studies.
The scientific team at ResolveMass Laboratories Inc. utilizes state-of-the-art analytical technologies, including LC-MS/MS, GC-MS, and ICP-MS platforms, to perform comprehensive extraction studies, validate analytical methods below established AET values, identify unknown chemical migrants, and conduct scientifically rigorous toxicological risk assessments.
By combining analytical excellence with extensive regulatory knowledge, ResolveMass Laboratories Inc. assists pharmaceutical organizations in navigating challenging FDA reviews, reducing development risks, and maintaining critical commercialization timelines.
For technical consulting, study design support, or regulatory assistance, developers may contact the scientific team at https://resolvemass.ca/contact/.
Frequently Asked Questions
Although both regulatory agencies prioritize patient safety, the FDA generally requires more extensive scientific justification and supporting evidence for E&L assessments. FDA reviewers often expect detailed analytical data, comprehensive toxicological evaluations, and clear explanations for decisions such as the selection of Uncertainty Factors. In contrast, the EMA may place greater emphasis on a risk-based approach, particularly when supported by established scientific rationale. The FDA is also more likely to require definitive identification of compounds detected above the Analytical Evaluation Threshold (AET).
No. The Safety Concern Threshold (SCT) is a toxicological exposure value that represents an acceptable daily intake level rather than an analytical reporting concentration. Before it can be applied within an E&L study, the SCT must be converted into an Analytical Evaluation Threshold (AET). This calculation incorporates factors such as maximum daily patient exposure, product dosage, packaging component characteristics, and analytical uncertainty. As a result, the AET becomes the appropriate reporting threshold for identifying and evaluating chemical migrants.
The required response timeline depends on the type of regulatory communication received from the FDA. Information Requests (IRs) issued during an active review cycle often require responses within a relatively short timeframe, sometimes as little as a few weeks. More significant deficiencies communicated through a Complete Response Letter (CRL) may require extensive laboratory work, toxicological assessments, or additional validation studies. In such cases, sponsors typically develop a broader resubmission strategy that may extend over several months.
The FDA evaluates single-use manufacturing components through a risk-based framework that considers the likelihood and extent of chemical migration into the product. Factors such as material composition, contact duration, process temperature, and exposed surface area are carefully reviewed. Regulators also examine whether downstream purification processes can effectively reduce or eliminate potential leachables before final product filling. This assessment helps determine whether manufacturing-related materials could impact product quality or patient safety.
Elastomeric components such as rubber stoppers, syringe plungers, and sealing systems contain complex mixtures of polymers and chemical additives. These materials often include curing agents, antioxidants, plasticizers, accelerators, and trace inorganic substances that may migrate into pharmaceutical formulations over time. Because they are commonly in direct and prolonged contact with drug products, they present a greater potential for chemical transfer. As a result, they require extensive extractables and leachables characterization during development.
The Expanded Decision Tree (EDT) serves as an advanced toxicological screening framework used to evaluate the potential hazards associated with chemical compounds identified during E&L studies. Unlike older classification systems, the EDT incorporates chemical structure and toxicological attributes to provide a more refined assessment of risk. This approach enables scientists to prioritize compounds requiring additional investigation and supports the establishment of scientifically justified exposure limits. Consequently, it has become a valuable tool for modern toxicological evaluations.
When sponsors rely on historical data or supplier-generated information, the FDA expects clear evidence that the data remain relevant to the current product and manufacturing process. This typically involves demonstrating material equivalency, comparable processing conditions, and consistent sterilization methods. Sponsors must also confirm that product formulations, exposure scenarios, and intended clinical use have not introduced new risks. Without adequate scientific justification, bridging arguments may not be accepted during regulatory review.
Compounds measured below the established AET are generally considered unlikely to present a meaningful toxicological concern and often do not require additional qualification. However, exceptions can occur when the compound belongs to a category of substances known to possess significant toxicity, such as potent mutagens or carcinogens. In these situations, the FDA may require targeted evaluation regardless of concentration. Therefore, compound identity and toxicological profile remain important considerations even when levels fall below the reporting threshold.
Reference:
- U.S. Food and Drug Administration. (2017, May 26). Complete response letter for NDA 208437 [FDA regulatory action letter]. U.S. Department of Health and Human Services. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208437Orig1s000OtherActionLtrs.pdf
- Kim, S., Jenke, D., Norwood, D. L., & Bowers, K. (2025). Uncertainty factors and relative response factors: Correcting detection and quantitation bias in extractables and leachables studies. PDA Journal of Pharmaceutical Science and Technology. Advance online publication. https://pmc.ncbi.nlm.nih.gov/articles/PMC12283826/
- Oktem, B., Nahan, K., Sussman, E. M., Yun, B. H., Patabandige, M. W., & Wickramasekara, S. (2021). Considerations for uncertainty factor determination in medical device extractables analysis [Scientific poster abstract]. U.S. Food and Drug Administration, Center for Devices and Radiological Health. https://www.fda.gov/science-research/fda-science-forum/considerations-uncertainty-factor-determination-medical-device-extractables-analysis
- U.S. Food and Drug Administration. (2025). Expanded decision tree: FDA’s food chemical toxicity screening tool. U.S. Department of Health and Human Services. https://www.fda.gov/food/food-chemical-safety/expanded-decision-tree-fdas-food-chemical-toxicity-screening-tool
- U.S. Food and Drug Administration. (2025, July 30). FDA releases new tool for toxicity screening of chemicals in food. U.S. Department of Health and Human Services. https://www.fda.gov/food/hfp-constituent-updates/fda-releases-new-tool-toxicity-screening-chemicals-food

