
Introduction
Generic Drug Development for Poorly Soluble APIs requires advanced Contract Development and Manufacturing Organization (CDMO) strategies to address the thermodynamic and kinetic limitations that restrict oral bioavailability. By utilizing sophisticated formulation technologies—including amorphous solid dispersions (ASDs), crystalline nanotechnology-based delivery systems, and lipid-based formulations (LBFs)—generic drug manufacturers can overcome dissolution barriers associated with solid-state drug substances while achieving pharmacokinetic performance comparable to the reference listed drug (RLD). Developing bioequivalent formulations for compounds categorized under Biopharmaceutics Classification System (BCS) Class II and Class IV remains one of the most technically demanding aspects of Abbreviated New Drug Application (ANDA) development. ResolveMass Laboratories Inc. supports these development programs through comprehensive solid-state characterization, reverse engineering, and advanced mass spectrometry, helping ensure both the physical integrity and chemical stability of drug products throughout the development lifecycle.
Learn more about executing successful comparative testing between generic and RLD products to establish bioequivalence.
Article Summary:
- Poor aqueous solubility remains a major barrier in generic drug development, particularly for BCS Class II and IV APIs, where inadequate dissolution can limit oral absorption and make bioequivalence with the reference listed drug (RLD) difficult to achieve.
- Amorphous Solid Dispersions (ASDs) enhance drug dissolution by converting crystalline APIs into stabilized amorphous forms within polymer matrices, creating supersaturation while minimizing crystallization through carefully selected carrier polymers and manufacturing processes.
- Nanocrystal technology improves bioavailability by reducing API particle size to the nanometer range, increasing surface area and dissolution rate without changing the drug’s crystalline structure, making it especially suitable for high-dose, poorly soluble compounds.
- Lipid-based formulations (LBFs) maintain highly lipophilic drugs in a dissolved state using oils, surfactants, and co-solvents, enabling improved intestinal absorption and, for suitable APIs, enhanced lymphatic transport that can reduce first-pass hepatic metabolism.
- Selecting the optimal formulation platform depends on multiple API-specific factors, including thermal stability, melting point, lipophilicity, dose requirements, solid-state properties, and manufacturability. Early formulation screening helps identify the most effective technology while reducing development risks.
- Advanced in vitro evaluation tools, including biorelevant dissolution media, dissolution-permeation studies, and physiologically based pharmacokinetic (PBPK) modeling, provide more accurate predictions of in vivo performance than conventional dissolution testing alone, improving confidence before clinical bioequivalence studies.
- A successful generic development strategy combines formulation innovation with comprehensive analytical characterization, including solid-state analysis, reverse engineering, bioanalytical testing, and stability assessment, to support robust ANDA submissions and increase the likelihood of first-cycle regulatory approval.
Formulation Engineering Challenges in Generic Drug Development for Poorly Soluble APIs
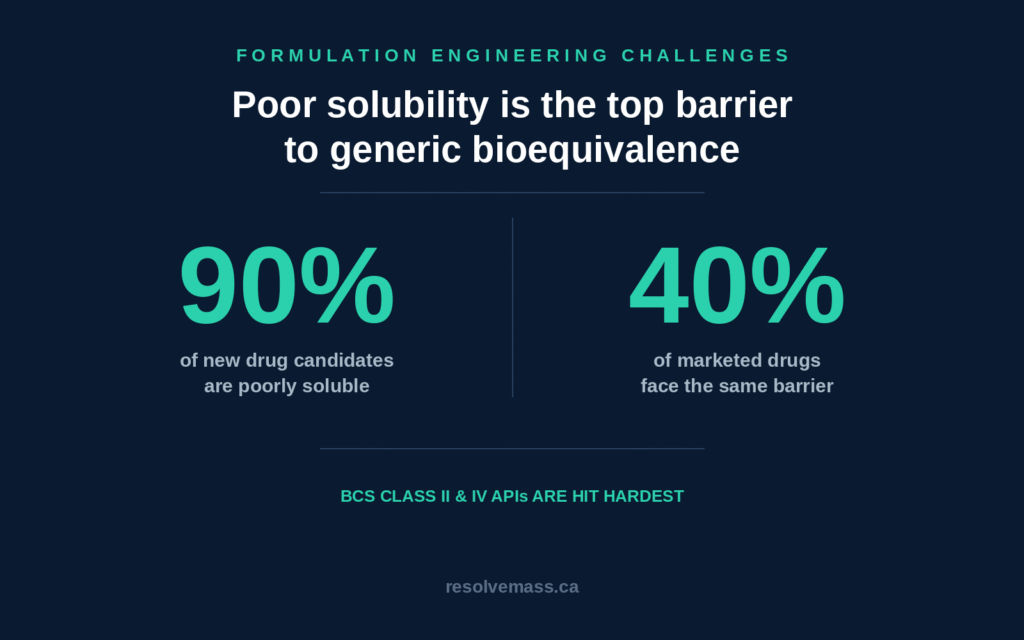
Designing generic formulations for poorly soluble active pharmaceutical ingredients requires a comprehensive understanding of thermodynamics, crystal lattice behavior, and the physicochemical mechanisms governing intestinal drug absorption. CDMOs must carefully investigate the intrinsic solid-state properties of each target molecule to overcome challenges such as slow dissolution, polymorphic conversion, and food-dependent variability in in vivo drug performance. Approximately 90% of investigational new chemical entities and nearly 40% of commercially available pharmaceutical products exhibit poor aqueous solubility, making this issue one of the most significant obstacles to achieving successful bioequivalence.
The fundamental challenge arises from thermodynamic stability. Crystalline APIs exist within highly ordered crystal lattices stabilized by strong intermolecular lattice forces. Dissolving these structures requires sufficient energy to disrupt the crystal lattice and solvate individual drug molecules within gastrointestinal fluids. In many cases, physiological conditions cannot provide enough energy to accomplish complete dissolution, resulting in poor absorption and inadequate systemic drug exposure. In addition, numerous hydrophobic compounds possess weak acidic or basic characteristics that exhibit pronounced pH-dependent solubility. Weakly basic drugs, for example, often dissolve readily in the acidic gastric environment but rapidly precipitate or recrystallize after entering the near-neutral or alkaline conditions of the intestine, producing inconsistent and highly variable pharmacokinetic behavior.
Polymorphic conversion during manufacturing processes—including wet granulation, milling, compression, or long-term storage—introduces another major formulation concern. An API initially present as a highly soluble metastable polymorph may gradually transform into a thermodynamically stable crystalline form with substantially lower solubility, leading to unexpected failures in dissolution testing and compromised product performance. To minimize these risks, formulation scientists rely on sophisticated analytical techniques for detailed solid-state characterization, including:
- Dynamic Vapor Sorption (DVS): Utilized to evaluate hygroscopic behavior, moisture-triggered crystallization, and transitions from amorphous to crystalline phases under carefully controlled relative humidity conditions.
- Inverse Gas Chromatography (iGC): Applied to characterize surface energy properties and investigate localized drug-excipient interactions, enabling prediction of physical compatibility prior to formulation optimization.
- X-ray Powder Diffraction (XRPD) and Modulated Differential Scanning Calorimetry (mDSC): Employed to assess crystallinity, confirm polymorphic identity, and determine glass transition temperatures (Tg).
Explore how advanced analytical testing prevents out-of-specification risks during the critical phase of manufacturing scale up for generic drugs.
Amorphous Solid Dispersions as a Key Solution
Amorphous solid dispersions (ASDs) improve drug solubility by molecularly dispersing a crystalline active pharmaceutical ingredient within a hydrophilic polymer matrix, thereby eliminating the high lattice energy associated with its crystalline form. This strategy transforms the API into a high-energy amorphous state capable of generating rapid supersaturation under physiological conditions, significantly increasing systemic drug exposure. The principal thermodynamic objective of an ASD is to achieve and maintain a supersaturated concentration of free drug that exceeds the equilibrium solubility of its crystalline counterpart, thereby creating a strong concentration gradient that promotes absorption across the intestinal epithelium.
Discover how solid-state analytics apply to complex molecular matrices in generic peptide and oligonucleotide projects.
Selection of Polymeric Carriers in Generic Drug Development for Poorly Soluble APIs
Choosing an appropriate polymer for amorphous solid dispersions depends on several critical parameters, including glass transition temperature (Tg), hygroscopic characteristics, and pH-responsive release behavior, all of which contribute to maintaining long-term kinetic stability during storage. Frequently utilized polymeric carriers include Hydroxypropyl Methylcellulose Acetate Succinate (HPMCAS), Copovidone, and Soluplus, each providing distinct stabilization mechanisms and dissolution characteristics. These polymers function by limiting the molecular mobility of the amorphous API, thereby suppressing crystal nucleation and preventing subsequent crystal growth.
HPMCAS is a versatile enteric, semi-synthetic cellulose derivative available in L, M, and H grades, distinguished by varying levels of acetyl and succinyl substitution. The L grade contains the highest proportion of succinyl groups, providing enhanced hydrophilicity and facilitating rapid drug release within the proximal small intestine. Conversely, the H grade possesses the greatest acetyl content, imparting greater hydrophobicity that slows drug release while offering exceptional inhibition of crystallization through hydrophobic adsorption onto the drug surface. HPMCAS also promotes sustained supersaturation by forming stable submicron colloidal structures within gastrointestinal fluids, thereby minimizing precipitation. However, processing HPMCAS using Hot-Melt Extrusion (HME) frequently requires temperatures above 170°C, potentially causing thermal degradation of both the polymer and heat-sensitive APIs unless suitable plasticizers are incorporated into the formulation.
Copovidone, a 6:4 copolymer composed of 1-vinyl-2-pyrrolidone and vinyl acetate (commercially available as Kollidon VA 64 or Plasdone S630), is widely selected because of its relatively low glass transition temperature of approximately 108–111°C and its pH-independent aqueous solubility. Incorporation of vinyl acetate reduces both hygroscopicity and polymer rigidity compared with homopolymeric PVP, resulting in excellent plasticity and processability during Hot-Melt Extrusion. Nevertheless, copovidone-based ASDs remain moderately hygroscopic. Moisture uptake during storage can substantially reduce the Tg of the dispersion, increasing molecular mobility and accelerating phase separation as well as API recrystallization over time. Soluplus is an amphiphilic graft copolymer specifically engineered for continuous hot-melt extrusion applications. It consists of a hydrophilic polyethylene glycol backbone grafted with vinyl caprolactam and vinyl acetate side chains. This specialized molecular architecture enables spontaneous micelle formation in aqueous environments, significantly improving dissolution performance.
Read about advanced characterization techniques utilized for complex therapeutics like liraglutide generic development services.
Manufacturing Platforms: Hot-Melt Extrusion versus Spray Drying
Hot-Melt Extrusion (HME) and Spray Drying (SD) are the two principal large-scale manufacturing technologies employed to produce stable amorphous solid dispersions. The choice between these platforms depends primarily on the thermal characteristics of the API, including melting point, thermal stability, and solubility in organic solvents. Although both technologies produce amorphous systems, they expose formulation components to fundamentally different thermodynamic environments during manufacturing.
HME is a continuous, solvent-free manufacturing technique that combines elevated temperatures with intense mechanical shear to dissolve the crystalline API directly within a molten polymer matrix exhibiting rubber-like properties. During processing, twin co-rotating screws continuously mix, knead, and disperse the formulation as it travels through a heated extrusion barrel. Successful processing requires maintaining melt viscosity within an optimized extrusion window ranging from approximately 1,000 to 10,000 Pa·s. Melt viscosities exceeding 10,000 Pa·s typically surpass the torque limitations of conventional extrusion equipment, whereas viscosities below 1,000 Pa·s produce materials that are excessively fluid for effective shaping through the extrusion die. Plasticizers, including polyethylene glycols (PEGs) and poloxamers, temporarily reduce the polymer’s Tg, permitting extrusion at lower temperatures while minimizing thermal degradation of temperature-sensitive APIs.
Spray Drying is a solvent-based manufacturing technology in which both the API and polymer are dissolved together in a volatile organic solvent, such as acetone, methanol, or dichloromethane, before being atomized through a spray nozzle into a stream of heated drying gas. Solvent evaporation occurs within milliseconds, rapidly increasing droplet viscosity and kinetically immobilizing drug molecules within a highly homogeneous amorphous matrix before crystallization can occur. Spray drying is particularly advantageous for APIs with high melting points or poor thermal stability and can be initiated using only milligram-scale quantities of drug substance. However, this technology requires substantial capital investment and involves multiple processing stages, including nitrogen inerting, solvent recovery infrastructure, and secondary drying operations to eliminate residual organic solvents.
Learn about analytical pathways and formulation tracking used for lanreotide generic development services.
Nanotechnology and Nanocrystalline Size Reduction Platforms
Nanocrystalline suspensions enhance dissolution by reducing crystalline drug particles into the submicron size range, generally between 100 and 500 nm, thereby dramatically increasing the available surface area for dissolution. Unlike amorphous systems, this carrier-free strategy preserves the thermodynamically stable crystalline structure of the API, eliminating concerns regarding recrystallization during storage. Nanosuspensions are especially appropriate for high-dose BCS Class IIa compounds where dissolution rate represents the primary limitation to oral bioavailability.
The improvement in dissolution kinetics achieved through nanosizing is fundamentally explained by the Noyes-Whitney equation. Decreasing particle diameter below one micrometer substantially increases the specific surface area (A) exposed to gastrointestinal fluids, thereby accelerating dissolution. Furthermore, according to the Prandtl boundary-layer theory, reducing particle size significantly decreases the thickness of the stagnant diffusion layer (h) surrounding each particle, facilitating faster drug diffusion into solution. When particle diameters decrease below approximately 100–200 nm, the Ostwald-Freundlich equation predicts an increase in saturation solubility (Cs) resulting from the high surface curvature and elevated dissolution pressure associated with nanocrystals. The combined effects of increased surface area and enhanced saturation solubility contribute to rapid drug dissolution, faster onset of therapeutic action, and highly reproducible absorption profiles.
Nanocrystalline systems are typically produced using either top-down mechanical techniques, including high-energy wet media milling and high-pressure homogenization, or bottom-up precipitation approaches. To inhibit particle agglomeration and prevent Ostwald ripening—a process in which smaller crystals dissolve and subsequently redeposit onto larger particles because of differences in surface chemical potential—the suspension must be stabilized using steric polymers such as HPC, HPMC, or PVP together with low concentrations of surfactants including sodium lauryl sulfate, Poloxamer 188, or lecithin. Converting these liquid nanosuspensions into stable solid oral dosage forms, including tablets and capsules, requires carefully controlled drying techniques for efficient moisture removal, as described in the following table.
Downstream Processing Methods for Nanocrystalline Suspensions
| Processing Method | Core Mechanism | Key Advantages | Primary Critical Quality Attributes (CQAs) & Challenges |
|---|---|---|---|
| Spray Drying | The nanosuspension is atomized into a heated drying gas stream, allowing rapid evaporation of water and formation of dry composite microparticles. | A continuous and scalable manufacturing process that produces free-flowing, spherical powders suitable for direct compression into tablets. | Elevated inlet temperatures may induce thermal degradation or irreversible fusion of nanoparticles. Careful optimization of inlet and outlet temperatures is essential. |
| Lyophilization (Freeze Drying) | The nanosuspension is frozen, followed by sublimation of ice through controlled primary and secondary vacuum drying cycles. | Eliminates thermal stress, making it particularly suitable for highly heat-sensitive or thermally labile active pharmaceutical ingredients. | Produces a highly porous cake that typically requires secondary milling. The process has relatively low throughput and high operational costs. |
| Spray-Freeze Drying | The nanosuspension is dispensed into liquid nitrogen, instantly freezing the droplets before sublimative freeze drying. | Produces highly porous, uniform spherical particles with excellent flow properties and rapid redispersion in water. | Requires specialized cryogenic processing equipment and precise optimization of protective polymers (such as HPC SSL or PVP) together with matrix-forming sugars. |
| Fluidized Bed Granulation | The nanosuspension is sprayed as a liquid binder onto fluidized carrier particles, such as sugars or sugar alcohols, where it dries to form a thin coating layer. | Integrates drying and granulation into a single, efficient manufacturing step while generating granules with excellent flowability. | The solubility of the carrier material directly influences nanoparticle release. Tight control of fluidization airflow, spray rate, and polymer concentration is necessary. |
Explore the structural control requirements underlying complex generic peptide semaglutide projects.
Lipid-Based Formulations for Lymphatic and Solubilization Pathways
Lipid-based formulations (LBFs) deliver highly lipophilic active pharmaceutical ingredients in a pre-solubilized state using carefully designed mixtures of oils, surfactants, and co-solvents, thereby eliminating the need for solid-state dissolution before absorption. Rather than relying on dissolution of crystalline drug particles within gastrointestinal fluids, this formulation strategy exploits the physiological lipid digestion process to maintain the drug in a stable mixed-micellar state throughout gastrointestinal transit. Because the drug remains solubilized during absorption, lipid-based formulations substantially reduce variability associated with different crystalline forms and food-dependent dissolution.
The Pouton Lipid Formulation Classification System divides lipid-based formulations into five distinct categories according to the quantitative proportions of triglyceride oils, lipophilic or hydrophilic surfactants, and water-miscible co-solvents. The primary objective of this classification is to optimize spontaneous self-emulsification and maintain efficient drug solubilization following exposure to aqueous gastrointestinal fluids. The structural characteristics and formulation behavior of each classification are summarized below.
Pouton Classification of Lipid-Based Formulations (LBFs)
| Formulation Type | Composition | Required Digestion? | Dispersion Droplet Size | Key Biopharmaceutical Advantages & Limitations |
|---|---|---|---|---|
| Type I | 100% pure triglycerides (medium-chain or long-chain lipids). | Yes. Pancreatic lipase digestion is required before dispersion occurs. | Coarse and highly variable dispersion. | Advantage: Excellent safety profile and strong solubilization capacity for highly lipophilic compounds. Limitation: Poor spontaneous dispersion and complete dependence on variable in vivo lipid digestion. |
| Type II | Lipids (40–80%) combined with lipophilic surfactants having HLB <10 (20–60%). | No. Undergoes spontaneous self-emulsification upon contact with water. | Emulsion droplets approximately 100–1000 nm. | Advantage: Rapid self-emulsification with good dilution stability. Limitation: Produces moderately sized droplets and generally requires sufficient gastrointestinal agitation for optimal dispersion. |
| Type IIIa (SMEDDS) | Lipids (40–80%), hydrophilic surfactants with HLB >10 (20–40%), and co-solvents (0–40%). | No. Forms spontaneous microemulsions. | Microemulsion droplets smaller than 100 nm with visually clear appearance. | Advantage: Excellent solubilization and rapid dispersion independent of gastric motility. Limitation: Elevated surfactant concentrations may contribute to gastrointestinal mucosal irritation. |
| Type IIIb (SNEDDS) | Lipids (<20%), hydrophilic surfactants (30–80%), and hydrophilic co-solvents (20–50%). | No. Produces spontaneous nanoemulsification. | Nanoemulsion droplets typically below 50 nm. | Advantage: Generates extremely fine nano-droplets with rapid absorption characteristics. Limitation: Increased likelihood of drug precipitation following extensive gastric dilution. |
| Type IV | Hydrophilic surfactants (30–80%) together with co-solvents (20–50%) without lipid content. | No. Readily miscible with aqueous media. | Micellar structures generally smaller than 20 nm. | Advantage: Provides maximum initial solubilization capacity for highly challenging drug molecules. Limitation: High risk of rapid precipitation following dilution and no contribution to lymphatic drug transport. |
The in vivo performance of lipid-based formulations is highly dependent on pancreatic lipase activity within the gastrointestinal tract. Pancreatic lipase enzymatically hydrolyzes triglycerides into free fatty acids and monoglycerides, which subsequently combine with endogenous bile salts and lecithin secreted from the gallbladder to form dynamic mixed micelles. These mixed-micellar structures efficiently transport dissolved drug molecules across the aqueous mucus layer and deliver them directly to the enterocyte membrane, thereby maximizing intestinal drug absorption.
For highly lipophilic APIs exhibiting log P values greater than 5, lipid-based formulations containing long-chain triglycerides (LCTs), including sesame oil or corn oil, provide an additional absorption pathway through the intestinal lymphatic system. Within enterocytes, digestion products of long-chain triglycerides are re-esterified into triglycerides and assembled into chylomicrons. Highly lipophilic drug molecules partition into these chylomicrons and are transported into intestinal lacteals rather than the portal circulation. Because lymphatic transport bypasses hepatic first-pass metabolism and enters systemic circulation through the thoracic duct, overall bioavailability increases substantially while reducing inter-patient pharmacokinetic variability.
Selecting the Optimal Solubility Enhancement Strategy
CDMOs determine the most appropriate solubility enhancement technology by aligning the unique solid-state characteristics and thermodynamic behavior of the API with manufacturing feasibility and development-stage objectives. Conducting parallel formulation screening using amorphous solid dispersions, nanosuspensions, and lipid-based systems enables generic drug developers to minimize development risks while identifying the formulation strategy most likely to achieve successful clinical bioequivalence. Selecting an unsuitable technology during early-stage formulation development may ultimately result in costly bioequivalence failures during clinical evaluation.
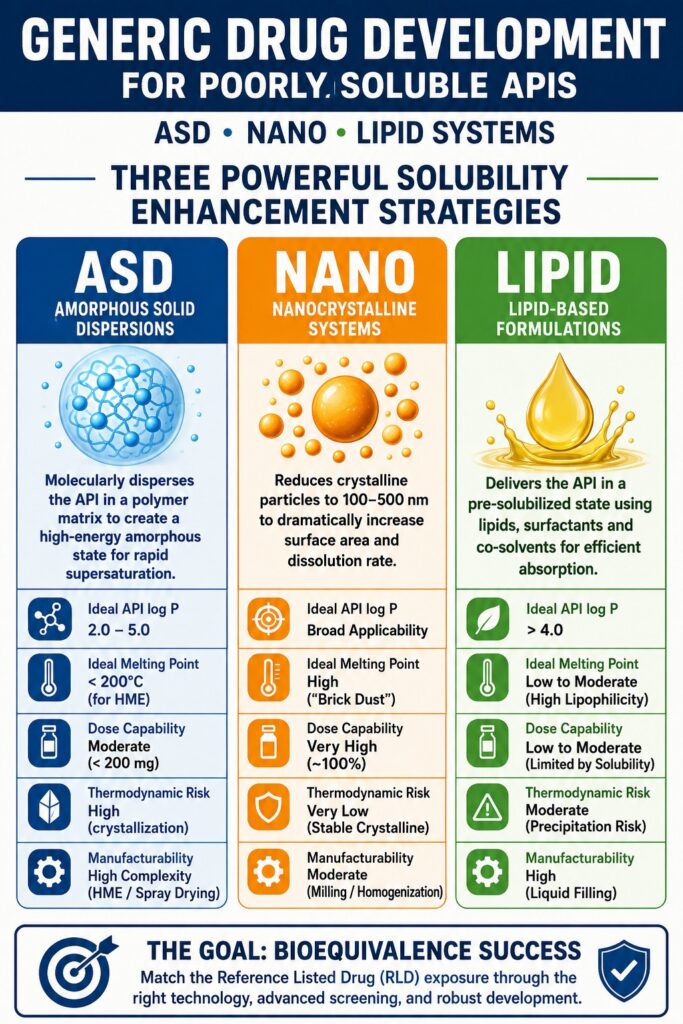
To facilitate systematic decision-making, the following comparison highlights the major characteristics that distinguish Amorphous Solid Dispersions, Nanocrystalline Systems, and Lipid-Based Formulations.
Comprehensive Technology Selection Matrix: ASD, Nano, and Lipid Systems
| Evaluation Parameter | Amorphous Solid Dispersions (ASD) | Nanocrystalline Systems | Lipid-Based Formulations (LBFs) |
|---|---|---|---|
| API Physical Form | Amorphous, molecularly dispersed or dissolved. | Crystalline submicron particles. | Fully solubilized within liquid lipid systems. |
| Ideal API log P | Moderate to high (2.0–5.0). | Broad applicability regardless of log P. | Very high, typically greater than 4.0. |
| Ideal API Melting Point | Low to moderate (Tm <200°C for HME processing). | High melting-point (“brick dust”) compounds. | Low to moderate melting point with strong lipophilicity. |
| Dose Capability | Moderate, generally below 200 mg because of drug-loading limitations. | Very high, with drug loading approaching 100%. | Low to moderate, constrained by lipid solubility. |
| Polymer Miscibility | Essential; high miscibility with the polymer carrier is required. | Not applicable. | Not applicable; requires sufficient solubility within the lipid phase. |
| Thermodynamic Risk | High potential for crystallization during manufacturing and storage. | Very low because the API remains in its stable crystalline state. | Moderate risk of precipitation after exposure to gastric fluids. |
| Lymphatic Transport | Low. | Low. | High when long-chain triglycerides are incorporated. |
| Manufacturability | Highly complex, requiring specialized HME or spray drying equipment. | Moderate complexity using conventional milling or homogenization technologies. | High manufacturability using standard soft-gel or hard-gel liquid filling equipment. |
Overcoming Bioequivalence Obstacles via Advanced In Vitro Screening
Successfully demonstrating clinical bioequivalence for poorly soluble drug products requires sophisticated in vitro screening approaches capable of reproducing the highly dynamic environment of the human gastrointestinal tract. Rather than depending exclusively on conventional USP dissolution testing, generic drug developers increasingly employ biorelevant dissolution media together with integrated dissolution-permeation models to obtain more accurate predictions of in vivo drug performance.
Traditional USP dissolution methods, including paddle and basket apparatuses, utilize relatively simple buffer systems containing synthetic surfactants such as sodium lauryl sulfate to establish sink conditions. Although these methods are highly valuable for routine quality control testing, they frequently overestimate drug solubility and fail to accurately reproduce physiological phenomena such as gastric precipitation, food-dependent absorption, and formulation-specific interactions within the gastrointestinal tract. Biorelevant dissolution testing addresses these limitations by employing simulated gastrointestinal fluids that closely mimic both fasted and fed physiological conditions.
Commonly used media, including FaSSIF and FeSSIF, contain physiologically relevant concentrations of bile salts such as sodium taurocholate together with lecithin, appropriate osmolarity, buffer capacity, and pH values representative of the stomach and proximal small intestine. These physiologically relevant media enable generic developers to evaluate formulation behavior during the transition from the acidic gastric environment to the more neutral intestinal environment, providing substantially improved prediction of in vivo dissolution performance.
Review validation protocols utilized to prove biosimilarity using LC-MS for advanced therapeutic equivalence workflows.
In addition, formulations containing high concentrations of surfactants—such as amorphous solid dispersions and lipid-based systems—may produce a phenomenon known as micellar entrapment, in which the API remains highly solubilized but is sequestered within surfactant micelles. Under these conditions, apparent solubility may increase substantially, while the thermodynamically active concentration of free drug available for intestinal membrane permeation actually decreases. Consequently, dissolution testing alone may significantly overestimate the true absorption potential of the formulation. To overcome this limitation, generic developers increasingly employ simultaneous dissolution-permeation (D-P) study designs.
These integrated analytical systems consist of a donor dissolution chamber containing biorelevant dissolution media separated from a receiver chamber by either a synthetic membrane, such as PAMPA, or a biological membrane model, including Caco-2 cell monolayers. The receiver chamber functions as a systemic sink while both dissolution and membrane permeation are monitored simultaneously. By independently quantifying drug dissolution and transmembrane permeation rates, scientists can distinguish the freely absorbable drug fraction from drug molecules that remain entrapped within micelles. This integrated approach provides a far more accurate prediction of in vivo absorption kinetics and enables identification of high-risk formulations before clinical bioequivalence studies begin.
Learn how multi-attribute screening supports comprehensive biosimilar characterization using mass spectrometry.
To further reduce development risks, CDMOs incorporate biorelevant experimental data into Physiologically Based Pharmacokinetic (PBPK) modeling. PBPK simulations utilize advanced computational algorithms to recreate human gastrointestinal physiology, including gastric emptying rates, intestinal transit times, luminal fluid volumes, and regional metabolic clearance. By incorporating dynamic non-sink dissolution and permeation profiles generated for both the generic formulation and the reference listed drug (RLD), PBPK models predict plasma concentration-time profiles, including Cmax and AUC, under simulated fasted and fed physiological conditions. These simulations enable formulation scientists to establish robust in vitro-in vivo correlations (IVIVC), define Clinically Relevant Specifications (CRS), optimize formulation variables, and maximize the likelihood of successful clinical bioequivalence and regulatory approval through the ANDA pathway.
ResolveMass Laboratories Inc. supports these advanced generic drug development programs by providing specialized analytical characterization and mass spectrometry services throughout formulation development. These capabilities include comprehensive reverse engineering and deformulation of the reference listed drug to establish its precise physicochemical profile, validated LC-MS/MS bioanalytical testing, and extractables and leachables (E&L) studies to confirm container-closure compatibility. In addition, the laboratory performs advanced solid-state characterization using Differential Scanning Calorimetry (DSC), Thermogravimetric Analysis (TGA), and high-field Nuclear Magnetic Resonance (NMR) spectroscopy to evaluate structural integrity, polymorphic purity, and thermodynamic behavior of enabling formulations. Integrating these comprehensive physicochemical and bioanalytical workflows significantly reduces regulatory risk while supporting faster and more reliable ANDA approvals.
Understand structural mapping methods by exploring how to utilize peptide mapping in biosimilars and complex generics.
Strategic Direction in Generic Drug Development for Poorly Soluble APIs
Successful Generic Drug Development for Poorly Soluble APIs depends on selecting the most appropriate physicochemical stabilization strategy and confirming formulation equivalence through predictive, biorelevant analytical evaluation. The implementation of advanced formulation technologies—including amorphous solid dispersions (ASDs), nanocrystalline delivery systems, and lipid-based formulations (LBFs)—allows generic manufacturers to overcome solubility limitations while closely replicating the in vivo exposure profile of the reference listed drug (RLD). Selecting the optimal technology based on the API’s solid-state characteristics, thermodynamic behavior, and biopharmaceutical properties is fundamental to achieving reliable bioequivalence and long-term product performance.
Mitigate formulation risks by scheduling comprehensive forced degradation of biosimilars and therapeutic small molecules.
Rather than relying exclusively on conventional dissolution methods that often fail to predict clinical outcomes, generic developers should adopt an integrated, multi-compartment evaluation strategy that simultaneously examines both drug solubility and membrane permeability. This comprehensive approach substantially reduces the risk of bioequivalence failure during clinical studies while providing greater confidence in formulation performance under physiological conditions. Comprehensive pre-formulation investigations, detailed deformulation of the reference product, and advanced solid-state characterization continue to serve as the foundation for successful first-cycle ANDA approvals. Collaborating with a specialized analytical laboratory and experienced CDMO partner such as ResolveMass Laboratories Inc. enables a streamlined progression from early formulation screening through process development and ultimately to commercial product launch.
Ensure long-term stability and regulatory readiness through targeted impurity profiling of biosimilars and complex generic drug assets.
To accelerate your generic drug development program and leverage specialized analytical testing, deformulation expertise, and advanced mass spectrometry capabilities, contact us today.
Frequently Asked Questions
The Ostwald-Freundlich equation explains how reducing particle size to the nanometer range increases the saturation solubility of a crystalline drug. As particle curvature becomes greater at extremely small sizes, the surface energy rises, making the drug dissolve more readily in biological fluids. This phenomenon not only accelerates dissolution but also improves drug availability for absorption, making nanocrystal technology particularly valuable for poorly soluble APIs.
Lipid-based formulations keep highly lipophilic drugs dissolved within a mixture of lipids, surfactants, and co-solvents before administration. After oral dosing, these systems rapidly form fine emulsions or mixed micelles that closely resemble the body’s natural lipid digestion process. Because the drug remains solubilized throughout gastrointestinal transit, absorption becomes more consistent under both fasted and fed conditions, thereby minimizing variability associated with food intake.
The limit of congruency refers to the maximum concentration of drug that can remain uniformly dispersed within a polymer matrix while dissolving together with the carrier. When this threshold is exceeded, the drug may separate from the polymer and create drug-rich regions on the dosage form surface. This separation reduces supersaturation, slows dissolution performance, and increases the likelihood of recrystallization during storage or after administration.
Traditional USP dissolution methods rely on simple buffer systems and synthetic surfactants that do not accurately reproduce the complex physiological conditions of the human gastrointestinal tract. These tests cannot fully capture important processes such as bile salt interactions, drug precipitation, micellar entrapment, or intestinal membrane permeation. As a result, formulations that appear acceptable during routine dissolution testing may still demonstrate poor or inconsistent in vivo bioavailability.
The Pouton Classification System organizes lipid-based formulations into five categories according to the proportions of oils, surfactants, and hydrophilic co-solvents they contain. Each formulation type exhibits different self-emulsification behavior, droplet size, and digestion characteristics after administration. This classification helps formulation scientists select the most appropriate lipid system for improving solubility, maintaining drug stability, and enhancing oral absorption of poorly soluble compounds.
Hydroxypropyl Methylcellulose Acetate Succinate (HPMCAS) stabilizes supersaturated drug solutions by limiting the formation and growth of drug crystals. Its amphiphilic molecular structure interacts with dissolved drug molecules and adsorbs onto developing crystal surfaces, preventing additional crystal growth. This mechanism helps maintain the drug in its amorphous, highly soluble state for longer periods, resulting in improved dissolution performance and enhanced oral bioavailability.
One of the most significant challenges is converting a liquid nanosuspension into a stable solid dosage form without causing irreversible nanoparticle aggregation. Drying techniques such as spray drying, freeze drying, and fluidized bed granulation must be carefully optimized to preserve particle size and maintain redispersibility. Stabilizing polymers and protective excipients are also essential to prevent crystal growth and ensure consistent product quality during storage.
The glass transition temperature (Tg) is a critical parameter that influences the physical stability of an amorphous solid dispersion. When storage temperatures approach or exceed the Tg, molecular mobility within the polymer matrix increases significantly, creating favorable conditions for drug recrystallization and chemical degradation. Maintaining an adequate Tg throughout the product’s shelf life helps preserve the amorphous structure, ensuring consistent dissolution behavior and long-term formulation stability.
Physiologically Based Pharmacokinetic (PBPK) modeling combines experimental dissolution and permeability data with mathematical representations of human physiology to predict in vivo drug exposure. These simulations estimate pharmacokinetic parameters such as Cmax and AUC under different physiological conditions before clinical studies are initiated. By identifying formulation risks early in development, PBPK modeling enables formulation optimization, reduces costly clinical failures, and improves the probability of achieving successful ANDA approval.
Reference:
- Li, M., Fan, Y., Zhou, Y., & Huang, Y. (2023). Strategies for improving the oral bioavailability of poorly water-soluble drugs. Pharmaceutics, 15(5), 1420. https://doi.org/10.3390/pharmaceutics15051420
- Sun, D., Lee, P. I., Hwang, K. M., Kim, H., Oh, D. H., Nguyen, K. H., Park, J. B., & Cui, J. H. (2020). Focusing on the formulation approaches of poorly water-soluble drug candidates for oral bioavailability enhancement: A review. Pharmaceutics, 12(7), 588. https://doi.org/10.3390/pharmaceutics12070588
- Karakitsios, E., Angelerou, M.-F.-G., Kapralos, I., Tsakiridou, G., Kalantzi, L., & Dokoumetzidis, A. (2025). Integrating in vitro dissolution and physiologically based pharmacokinetic modeling for generic drug development: Evaluation of amorphous solid dispersion formulations for tacrolimus. Pharmaceutics, 17(2), 227. https://doi.org/10.3390/pharmaceutics17020227
- Awasthi, A., & Gupta, M. (2026). Generic drug development and bioequivalence study. International Journal of Pharmaceutical Sciences, 4(6), 369–391. https://doi.org/10.5281/zenodo.20492371
- Daalmann, M., Kimmel, V., Muehlenfeld, C., Thommes, M., & Winck, J. (2025). Characterization of different copovidone grades as carrier materials in hot melt extrusion of amorphous solid dispersions. Pharmaceutics, 17(9), 1138. https://doi.org/10.3390/pharmaceutics17091138
- He, Y., & Ho, C. (2015). Amorphous solid dispersions: Utilization and challenges in drug discovery and development. Journal of Pharmaceutical Sciences, 104(10), 3237–3258. https://doi.org/10.1002/jps.24541

