
Global Market Dynamics and Generic Injectable Trends
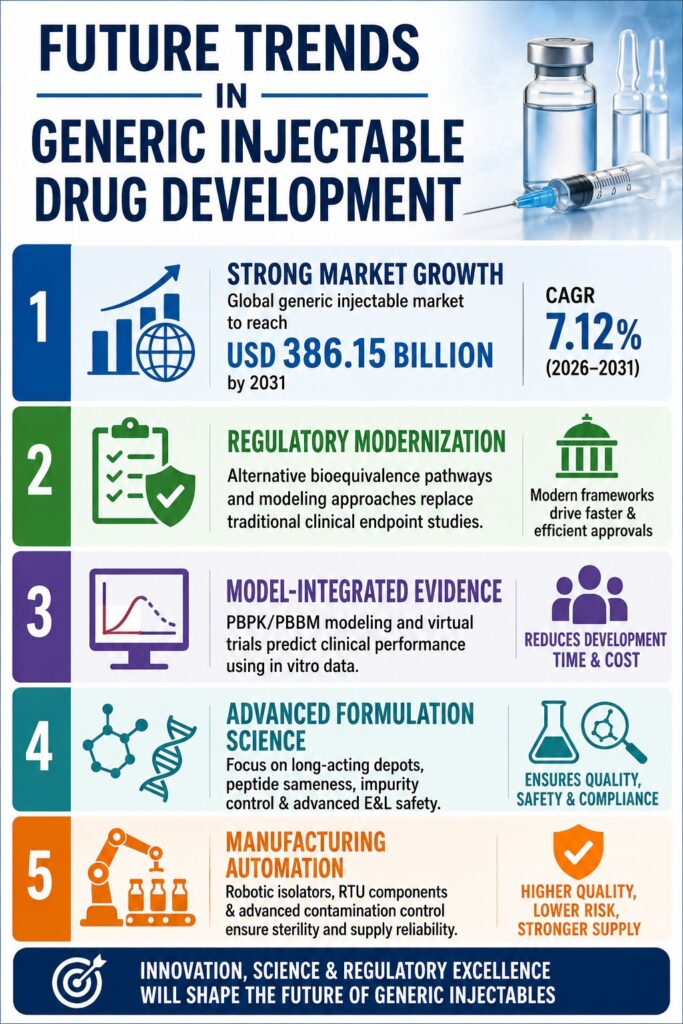
The global market for sterile injectables is undergoing a significant transformation, rapidly shifting from highly commoditized small-molecule formulations toward advanced, long-acting, and complex parenteral therapies. This clinical and manufacturing evolution, which is collectively shaping modern Generic Injectable Trends, is expected to drive the global market to USD 386.15 billion by 2031, reflecting a stable compound annual growth rate (CAGR) of 7.12%.
To succeed in this market, developers must master complex drug delivery systems. Learn more about our generic peptide semaglutide projects.
This long-term market expansion is being driven by a convergence of several critical factors, including major patent expirations, increasing prevalence of chronic diseases worldwide, and intensified cost-containment efforts by public healthcare systems and insurance providers. Originator biologics and complex peptide therapeutics accounting for more than USD 100 billion in annual sales are anticipated to lose patent protection between 2024 and 2028. The rapid adoption of biosimilars clearly demonstrates this trend. For instance, eight adalimumab biosimilars secured approximately 35% of the United States volume share within eighteen months following the loss of exclusivity of the reference product.
Explore our specialized services for generic peptide oligonucleotide projects.
Furthermore, policy initiatives are accelerating generic adoption across global healthcare systems. Germany’s implementation of automatic biosimilar substitution in early 2025, combined with centralized procurement programs such as Brazil’s BRL 2.1 billion (USD 420 million) award for oncology and insulin injectables in 2025, illustrates how governmental actions are facilitating broader integration of generic products. In the United States, Medicare price negotiations introduced under the Inflation Reduction Act beginning in 2026 are expected to further increase the price differential between branded medicines and generic injectables, creating a strong economic incentive for healthcare providers and patients to transition toward bioequivalent alternatives.
| Market Forecast Segment | 2025 Value (USD) | 2026 Value (USD) | Projected Future Value (USD) | Forecast Period / CAGR | Primary Sector Drivers |
|---|---|---|---|---|---|
| Global Generic Injectable Market | 255.59 Billion | 273.78 Billion | 386.15 Billion by 2031 | 2026–2031 / 7.12% CAGR | Peptide hormone patent expirations, biosimilar insulins, monoclonal antibody biosimilars |
| U.S. Generic Sterile Injectable Market | 17.91 Billion | 19.73 Billion | 43.26 Billion by 2035 | 2026–2035 / 9.22% CAGR | Oncology therapeutics, chronic disease management, advanced drug-device systems |
| Global Sterile Injectable Drugs Market | 593.00 Billion | 640.70 Billion | 1.12 Trillion by 2035 | 2026–2035 / 8.20% CAGR | Rapid expansion of oncology immunotherapies, monoclonal antibodies, and biologics CDMO outsourcing |
| Global Sterile Injectables CDMO Market | 37.82 Billion | 42.06 Billion | 87.34 Billion by 2033 | 2026–2033 / 11.20% CAGR | Outsourcing of complex biomanufacturing, gene therapies, and aseptic fill-finish operations |
| U.S. Specialty Injectable Generics Market | 24.29 Million | 26.35 Million | 39.58 Million by 2031 | 2026–2031 / 8.48% CAGR | Transition toward technology-driven complex formulations including liposomes, long-acting depots, and peptides |
Article Summary:
- The global generic injectable market is expected to reach USD 386.15 billion by 2031, driven by patent expirations, chronic disease growth, and healthcare cost-saving efforts.
- Demand is shifting from traditional injectables to complex products such as biosimilars, peptides, and long-acting formulations.
- Regulatory agencies are simplifying approvals through alternative bioequivalence methods, reducing reliance on large clinical trials.
- Computational modeling and virtual trials are helping manufacturers speed up development and lower costs.
- Long-acting injectable products require advanced material science and precise formulation matching to ensure equivalent performance.
- Peptide generics need rigorous structural characterization and impurity control to meet safety and regulatory standards.
- Future success will depend on advanced manufacturing, automation, strong regulatory strategies, and specialized analytical expertise.
Regulatory Modernization and Alternative Bioequivalence Pathways in Generic Injectable Trends
Regulatory authorities worldwide are actively modernizing approval frameworks to support the development of complex generic products through alternative bioequivalence pathways based on advanced analytical characterization. This evolving regulatory environment, strengthened through GDUFA III in the United States and aligned initiatives within Health Canada, enables manufacturers to replace costly clinical endpoint studies with robust in vitro and in silico evaluations.
Leverage expert assistance for lanreotide generic development services.
Within the United States, the USFDA Office of Generic Drugs leverages its regulatory science initiatives to issue Product-Specific Guidances (PSGs). These documents outline the agency’s recommendations for demonstrating bioequivalence to designated Reference Listed Drugs (RLDs). The release of a finalized PSG serves as an important market catalyst, with research indicating that availability of an active PSG increases the likelihood of an Abbreviated New Drug Application (ANDA) submission by approximately 3.8 times.
Optimize your regulatory pathway with liraglutide generic development services.
To improve first-cycle approval rates, which have historically ranged between 9% and 15%, the GDUFA III pre-submission meeting program enables developers to engage in early scientific discussions with regulators regarding proposed alternative bioequivalence methodologies. Under this framework, bioequivalence assessments increasingly rely on a comprehensive “weight-of-evidence” approach that integrates in vitro release testing (IVRT), in vitro permeation testing (IVPT), and detailed physicochemical characterization rather than relying exclusively on traditional clinical endpoint studies.
Health Canada maintains a similar regulatory structure under the Food and Drug Regulations. Generic manufacturers seeking market authorization must submit an Abbreviated New Drug Submission (ANDS) to obtain both a Notice of Compliance (NOC) and a Drug Identification Number (DIN). To establish bioequivalence, Health Canada requires that the 90% confidence interval of the relative mean area under the curve (AUC) of the test product compared with the Canadian Reference Product (CRP) falls within the accepted statistical range of 80% to 125%. For narrow therapeutic index products and modified-release formulations, requirements become more stringent, requiring the 90% confidence interval of the relative mean maximum concentration (Cmax) to also remain within specified limits under both fed and fasted conditions.
[
\text{90% CI of } \left( \frac{\text{Test } AUC}{\text{Reference } AUC} \right) \in [80%, 125%]
]
and
[
\text{90% CI of } \left( \frac{\text{Test } C_{max}}{\text{Reference } C_{max}} \right) \in [80%, 125%]
]
Health Canada ANDS Regulatory Approval Pipeline
[ ANDS Submission ] -> [ HPFB Scientific Review ] -> [ NOC Database Listing ] -> [ DIN Issued ]Health Canada has also proposed amendments that would permit generic products containing different physicochemical forms, including salts, esters, or complexes, of the same active moiety to proceed through the ANDS pathway. Although the proposal aims to improve patient access to affordable medications, it has generated debate within the pharmaceutical industry. Critics argue that alternative salt or ester forms represent distinct chemical entities with unique physicochemical and stability characteristics that may influence localized safety and therapeutic performance.
As a result, manufacturers must generate extensive analytical evidence demonstrating that any physicochemical differences do not produce altered degradation behavior or significantly different impurity profiles. Early regulatory engagement combined with advanced analytical characterization remains essential for ensuring that generic formulations comply with both international standards and evolving regional regulatory expectations.
Mathematical Modeling and Virtual Trials in Generic Injectable Trends
The incorporation of computational modeling and simulation into generic drug development is establishing a new benchmark for demonstrating bioequivalence without relying exclusively on conventional human clinical trials. These approaches, collectively referred to as Model-Integrated Evidence (MIE), employ sophisticated in silico methodologies and virtual crossover studies to predict clinical performance using in vitro physicochemical data.
Under GDUFA III, the USFDA’s adoption of Quantitative Methods and Modeling (QMM) is transforming regulatory decision-making processes. Several collaborative initiatives have been introduced, including the Model Master File (MMF) pilot program, which enables modeling experts to submit validated computational frameworks that may be referenced across multiple regulatory applications. This strategy supports Model-Integrated Bioequivalence (MIBE) by combining principles from clinical pharmacology, biopharmaceutics, and materials science.
Model-Integrated Evidence (MIE) Workflow
[ In Vitro Physicochemical Data ] ---> [ Local PBPK / PBBM Model ] ---> [ Virtual Crossover Trial ]
|
[ Alternative Bioequivalence ] <--- [ Model Validation via PopPK ] <-----------+A foundational component of the MIE framework involves physiologically based pharmacokinetic (PBPK) and physiologically based biopharmaceutics modeling (PBBM). PBPK models integrate human physiological characteristics, including tissue volumes, blood flow patterns, and interstitial fluid dynamics, with formulation-specific physicochemical attributes. For sterile injectables, localized PBPK models simulate drug absorption and matrix degradation directly at the administration site, including subcutaneous and deep intramuscular tissues.
These computational tools facilitate virtual bioequivalence (VBE) studies conducted within simulated patient populations. By evaluating hundreds of virtual crossover scenarios, developers can assess clinical risks across a wide range of physiological conditions:
- Dissolution Profile Deviations: Determining whether minor differences in release rates among manufacturing batches affect in vivo therapeutic exposure.
- Alcohol-Induced Dose Dumping: Assessing whether localized exposure to co-solvents may trigger premature release in extended-release formulations.
- Locally Acting Site-of-Action Dynamics: Predicting tissue-specific drug concentrations for complex products where systemic plasma levels are poor indicators of therapeutic activity.
- Population Covariate Impact: Evaluating how patient-specific factors such as subcutaneous fat thickness, tissue vascularization, and age-related physiological changes influence pharmacokinetic outcomes.
To address challenges associated with incomplete pharmacokinetic datasets, population pharmacokinetic (PopPK) modeling is increasingly utilized for individual-level data imputation. During long-term pharmacokinetic studies, PopPK models can estimate missing blood sampling data, preserving study integrity.
A notable regulatory approval in 2023 demonstrated the value of this approach, where validated PopPK-based data imputation served as the primary bioequivalence evidence supporting a complex generic ANDA. This strategy prevented invalidation of the clinical study and highlighted the growing role of predictive modeling in regulatory submissions. Through the integration of in vitro characterization and advanced computational algorithms, generic manufacturers can reduce development timelines by several months to more than two years while achieving substantial research and development cost efficiencies.
Technological Drivers and Generic Injectable Trends in Long-Acting Depots
Material Science and Biodegradable Matrices
Long-acting injectable depots provide sustained therapeutic delivery by incorporating active pharmaceutical ingredients into biodegradable, specially engineered polymeric matrices. To compete successfully within this high-value market segment, generic manufacturers must demonstrate precise Q1, Q2, and Q3 microstructural sameness to replicate the degradation characteristics and release kinetics of the reference product.
Controlled-release long-acting injectables (LAIs) frequently utilize advanced biodegradable polymers such as Poly(lactic-co-glycolic acid) (PLGA), Poly(lactic acid) (PLA), and Poly(lactide-co-caprolactone) (PLCL). These polymers undergo degradation in vivo through random hydrolytic cleavage of ester bonds, a process highly dependent upon their microstructural composition. Three critical material attributes govern this degradation behavior:
Copolymer Ratio (Lactide-to-Glycolide)
Glycolic acid units possess greater hydrophilicity than lactic acid units. Increasing glycolide content accelerates water penetration into the polymer matrix, enhancing hydrolytic degradation and increasing drug release rates.
Molecular Weight Distribution (Mw/Mn)
Average molecular weight (Mw) and the polydispersity index (Mw/Mn) directly influence mechanical properties, glass transition temperature (Tg), and matrix erosion behavior.
Polymer End-Group Chemistry
Polymers containing free carboxylic acid end groups exhibit greater hydrophilicity and faster hydrolytic degradation. In contrast, ester-capped polymers demonstrate reduced water uptake and more prolonged degradation profiles.
PLGA Hydrolytic Cleavage Pathway (Sustained Release Mechanism)
O O O O
|| || || ||
--[ O-CH-C ]x -- [ O-CH2-C ]y -- [ O-CH-C ]x -- [ O-CH2-C ]y --
| |
CH3 CH3
(Lactide) (Glycolide) (Lactide) (Glycolide)
|
v + H2O (Hydrolysis)
Lactic Acid + Glycolic Acid (Metabolic Clearance)For generic developers, achieving exact Q3 microstructural equivalence remains particularly challenging. The formulation’s final thermodynamic properties, viscosity, pore architecture, and particle size distribution are highly sensitive to processing variables including shear forces, solvent evaporation kinetics, and drying methodologies.
Ensure regulatory success with biosimilar characterization using mass spectrometry.
ResolveMass Laboratories Inc. assists developers in overcoming these challenges through specialized analytical characterization and custom polymer synthesis capabilities. Supported by PhD-level scientific expertise, the laboratory employs high-resolution Gel Permeation Chromatography (GPC) coupled with multi-angle light scattering detectors to accurately characterize molecular weight distributions.
In addition, ResolveMass utilizes quantitative nuclear magnetic resonance (qNMR) and Fourier-Transform Infrared (FTIR) spectroscopy to verify copolymer composition and end-group chemistry. These analytical capabilities support reverse engineering of innovator formulations, selection of suitable development candidates, and achievement of regulatory compliance.
Validate your product’s efficacy through comparative testing between generic and RLD.
Peptide Sameness and Impurity Profiling Challenges
Demonstrating therapeutic equivalence for synthetic peptide injectables requires extensive physicochemical characterization of the active pharmaceutical ingredient together with comprehensive impurity control strategies. Regulatory agencies require manufacturers to establish complete structural identity relative to the reference product while demonstrating that synthetic impurities do not increase immunogenicity risk.
Peptide therapeutics, defined as polymers consisting of 40 or fewer amino acids, occupy a unique regulatory category between traditional small molecules and large biologic proteins. Peptides produced through recombinant DNA (rDNA) technology generally do not qualify for conventional ANDA pathways. However, advances in solid-phase peptide synthesis (SPPS) and liquid-phase peptide synthesis (LPPS) have enabled generic manufacturers to produce synthetic versions of both recombinant and synthetic innovator peptides, including liraglutide, semaglutide, and teriparatide.
Utilize our advanced peptide mapping in biosimilars to ensure quality.
To establish API sameness under USFDA synthetic peptide guidance, generic products must demonstrate equivalence across five primary physicochemical dimensions:
- Primary Sequence Mapping: Confirmation of the exact amino acid sequence and stereochemical configuration.
- Secondary and Tertiary Structure: Characterization of alpha-helical, beta-sheet, and random coil conformations using circular dichroism, FTIR, and NMR techniques.
- Oligomerization and Aggregation State: Evaluation of self-association tendencies that may contribute to immunogenic responses.
- Physicochemical Properties: Assessment of molecular charge, isoelectric point (pI), and chromatographic retention characteristics.
- Biological Bioactivity: Functional testing through cell-based assays to confirm equivalent biological performance.
Peptide ANDA Impurity Threshold and Action Requirements
| Concentration | Action Required |
| ≥ 0.10% | Must be identified and structurally characterized. Must be controlled below or equivalent to RLD levels. |
| 0.10% – 0.50% | New or elevated impurities require scientific justification demonstrating no increase in immunogenicity risk. In silico and in vitro T-cell evaluations are required. |
| > 0.50% | Unacceptable. Process optimization is required. |
Beyond API sameness, manufacturers must rigorously control peptide-related impurities. Chemical synthesis processes may generate impurities including amino acid deletions, insertions, incomplete deprotections, diastereomers, and beta-isomerization products. Such impurities may alter immunogenic profiles and increase the potential for adverse immune responses.
Accordingly, any peptide-related impurity present at concentrations of 0.10% or greater must be formally identified and characterized. When a novel or elevated impurity falls between 0.10% and 0.50%, manufacturers must demonstrate that the impurity does not increase immunogenicity risk through integrated in silico assessments and in vitro biological testing.
| Impurity Risk Assessment Phase | Core Methodologies | Analytical and Clinical Parameters | Regulatory Deliverables |
| Phase I: Sequence Analysis | In silico human homology mapping | EpiMatrix and JanusMatrix algorithms; HLA-binding prediction | Predicts T-cell epitopes and identifies sequence variations |
| Phase II: Physical Chemistry | High-resolution mass spectrometry | LC-MS/MS (Q-Exactive Orbitrap), multidimensional qNMR | Resolves co-eluting impurities and characterizes structural modifications |
| Phase III: Immunogenicity | In vitro cellular assays | PBMC activation assays; T-cell proliferation profiles | Quantifies immunogenicity risk and supports impurity specifications |
Assess your products with our post-translational modifications (PTMs) in biosimilars expertise.
To support these advanced programs, ResolveMass Laboratories Inc. provides specialized peptide characterization services. Using high-resolution LC-MS/MS platforms, including the Q-Exactive Orbitrap, together with multidimensional NMR spectroscopy, the laboratory performs detailed peptide sequence mapping, secondary structure evaluation, and trace-level impurity characterization. These services assist developers in preparing robust DMF submissions and meeting stringent regulatory expectations.
Advanced Impurity Controls Under Evolving ICH Q3E and Nitrosamine Guidelines
The chemical safety of generic sterile injectables is governed by comprehensive risk assessment programs designed to identify and control trace-level migratory contaminants and mutagenic compounds. The emerging ICH Q3E guideline is expected to harmonize global expectations for extractables and leachables, while regulatory authorities continue enforcing strict limits for carcinogenic nitrosamine impurities.
Parenteral formulations interact with numerous materials throughout manufacturing, storage, transportation, and administration. These include elastomeric syringe components, single-use bioreactor systems, primary packaging materials, and administration devices. Such interactions create opportunities for chemical migration into drug products.
Extractables are compounds that can be released from contact materials under exaggerated laboratory extraction conditions involving elevated temperatures, prolonged contact periods, or aggressive solvents.
Leachables are compounds that actually migrate into the drug product under normal manufacturing, storage, and usage conditions.
The draft ICH Q3E guideline, endorsed as Step 2 in August 2025 and anticipated for finalization in June 2027, establishes a globally harmonized framework for extractables and leachables (E&L) risk assessment grounded in ICH Q9 quality risk management principles. The guideline requires calculation of an Analytical Evaluation Threshold (AET) for each finished drug product container-closure system. Any compound detected above this threshold must be identified, quantified, and toxicologically assessed.
The AET is calculated according to the following equation:
[
\text{AET} = \frac{\text{SCT} \times D_v}{D_d \times U_f}
]
Where:
- SCT = Safety Concern Threshold (typically 1.5 µg/day based on lifetime mutagenic and carcinogenic exposure considerations under ICH M7).
- Dv = Volume of extraction solvent utilized during the study (mL).
- Dd = Maximum daily patient dose (mL/day).
- Uf = Analytical uncertainty factor (typically 1.5–4.0).
Alongside E&L evaluations, regulatory agencies require strict monitoring of nitrosamine impurities. Nitrosamines such as N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) are potent mutagenic carcinogens. These compounds may form when secondary or tertiary amines react with nitrite sources under acidic conditions during API manufacturing or through interactions with packaging materials and contaminated water systems.
Regulatory agencies have established extremely low acceptable intake limits, including 96 ng/day for NDMA and 26.5 ng/day for NDEA. Consequently, highly sensitive analytical methods are required to demonstrate compliance throughout the product lifecycle.
ResolveMass Laboratories Inc. supports these requirements through dedicated E&L and nitrosamine testing services. Operating from a USFDA-registered facility with an ISO 9001:2015 certified quality management system, the laboratory develops custom validated methods using High-Resolution LC-MS/MS platforms, including the Q-Exactive Orbitrap, as well as Triple Quadrupole (QqQ) LC-MS instrumentation.
These advanced technologies enable accurate identification of unknown compounds while achieving ultra-low detection capabilities, including Limits of Detection (LOD) as low as 0.03 ng/mL and Limits of Quantification (LOQ) of 0.1 ng/mL. Such capabilities assist manufacturers in maintaining product safety and meeting FDA and Health Canada requirements.
Manufacturing Automation and Sterility Assurance Trends
Sterile manufacturing operations are rapidly transitioning from traditional manual cleanroom environments toward highly automated, robotic, and isolated production systems designed to maximize sterility assurance and minimize contamination risk. These advancements are essential for reducing human error and addressing manufacturing quality challenges that have historically contributed to shortages of generic sterile medicines.
Because parenteral products are administered directly into systemic circulation, bypassing the body’s natural protective barriers, even minimal microbial or particulate contamination can result in severe patient harm. While many small-molecule injectables can undergo terminal sterilization using heat, steam, or gamma irradiation after filling, complex products such as peptides, monoclonal antibodies, and liposomal formulations often require aseptic processing throughout manufacturing.
Aseptic Containment Architecture Comparison
Traditional Grade A Cleanroom:
[ Humans in Sterile Gowns ] ---> [ Open Fill Line ] ---> [ High Human Interruption Risk ]
Advanced Robotic Isolator:
[ Robotic Manipulation ] ---> [ Sealed Class Barrier Enclosure ] ---> [ VHP Decontamination ]To optimize sterile manufacturing performance, facilities are increasingly implementing advanced containment technologies:
Restricted Access Barrier Systems (RABS) and Isolators
Closed isolator systems physically separate personnel from aseptic processing zones. Automated vaporized hydrogen peroxide (VHP) decontamination cycles maintain Grade A conditions within the filling environment.
Robotic Fill-Finish Automation
High-speed robotic systems perform critical operations including vial de-nesting, filling, and stopper placement. Integrated sensors and non-destructive weight verification systems help maintain dosing accuracy and improve yield.
Ready-to-Use (RTU) Components
Modern facilities increasingly utilize pre-washed, pre-depyrogenated, and pre-sterilized vials, syringes, and cartridges supplied in ready-to-use formats. Adoption of RTU components eliminates the need for extensive washing and depyrogenation infrastructure, reducing facility footprint and energy requirements.
Contamination Control Strategies (CCS)
Comprehensive CCS programs integrate environmental monitoring, particulate surveillance, and non-destructive Container Closure Integrity (CCI) testing methods such as vacuum decay and high-voltage leak detection to verify package integrity.
These automation initiatives also play a critical role in reducing persistent global drug shortages. Sterile injectables account for approximately two-thirds of active pharmaceutical shortages worldwide. The generic marketplace is characterized by significant pricing pressure resulting from purchasing organizations such as Vizient, Premier, HPG, Red Oak, and ClarusOne.
Compressed profit margins often limit investment in manufacturing upgrades. Consequently, when regulatory inspections identify contamination events, particulate issues, equipment failures, or compliance deficiencies at major facilities, production interruptions can significantly impact national supply.
Because sterile injectable manufacturing requires highly specialized infrastructure and many competing manufacturers already operate near capacity, alternative suppliers are often unable to rapidly compensate for lost production. The resulting supply disruptions may persist for extended periods.
By implementing robotic isolators, RTU technologies, and advanced contamination control systems, manufacturers can enhance batch consistency, reduce contamination-related shutdown risks, and strengthen long-term supply chain resilience.
Strategic Horizon for Generic Injectable Trends
Achieving leadership within the sterile parenteral marketplace requires a comprehensive development strategy that combines advanced formulation science, computational bioequivalence modeling, robust analytical characterization, and proactive regulatory planning. Manufacturers capable of successfully navigating these technical barriers will be positioned to establish sustainable competitive advantages and capture long-term commercial opportunities.
As patent exclusivity continues to expire for major biologics and peptide therapeutics, ongoing commoditization of traditional small-molecule injectables will continue to intensify pricing pressures. Future growth within the generic injectable sector will increasingly depend on complex generics, where sophisticated manufacturing processes, analytical requirements, and regulatory barriers naturally limit competition.
Success in this evolving environment requires deep scientific expertise, advanced characterization technologies, and a forward-looking regulatory strategy. Collaborating with specialized contract research organizations can provide access to the technical resources and regulatory support necessary to accelerate development timelines and reduce program risk.
ResolveMass Laboratories Inc. serves as a strategic partner for developers operating within this rapidly evolving landscape. Combining PhD-level scientific expertise with advanced polymer synthesis capabilities, peptide characterization platforms, and highly sensitive mass spectrometry workflows, the organization delivers scientifically rigorous and regulatory-compliant data packages that support successful submissions.
Whether supporting custom polymer development, peptide sequence characterization, extractables and leachables studies, or advanced nitrosamine testing programs, ResolveMass provides the analytical depth required to advance complex generic injectable pipelines efficiently and confidently.
Contact us to discuss your technical challenges and accelerate your development timeline.
Frequently Asked Questions
The global generic sterile injectable market continues to show strong growth due to increasing demand for affordable injectable therapies and the expanding use of biosimilars. The market was valued at USD 255.59 billion in 2025 and is expected to reach USD 273.78 billion in 2026. Industry forecasts indicate that the market could grow to approximately USD 386.15 billion by 2031, supported by a projected CAGR of 7.12%. Rising chronic disease prevalence and patent expirations are key contributors to this expansion.
Aseptic filling is a manufacturing process in which sterilized drug products and packaging components are brought together under highly controlled sterile conditions. This approach is commonly used for products that cannot tolerate heat or radiation. Terminal sterilization, on the other hand, occurs after the product has been filled and sealed, exposing the final packaged product to a sterilization process such as steam, heat, or irradiation. Both methods aim to ensure sterility, but they differ significantly in their operational approach.
The Analytical Evaluation Threshold (AET) is calculated using a formula that incorporates the Safety Concern Threshold (SCT), extraction volume, maximum daily patient dose, and an analytical uncertainty factor. This value serves as a scientifically justified limit for evaluating extractables and leachables. Any compound detected above the calculated threshold must undergo identification, quantification, and toxicological assessment. The AET helps manufacturers establish a consistent risk-based approach for product safety evaluations.
Synthetic peptides possess structural and functional characteristics that differ significantly from conventional small-molecule drugs. Their amino acid sequences, folding patterns, aggregation behavior, and potential immunogenicity require more specialized assessment methods. Because of these complexities, standard impurity guidelines such as ICH Q3A and Q3B are not considered sufficient. Instead, peptide products are evaluated using dedicated regulatory frameworks that focus on structural equivalence, impurity characterization, and biological activity.
Demonstrating peptide API sameness requires a comprehensive analytical strategy that examines multiple structural and functional attributes of the molecule. High-resolution mass spectrometry is used to confirm amino acid sequence integrity, while multidimensional nuclear magnetic resonance helps characterize higher-order structures. Additional techniques evaluate aggregation behavior and physicochemical properties. Cell-based bioassays are also employed to confirm that the biological activity of the generic peptide matches that of the reference product.
Ongoing shortages of generic sterile injectables are often linked to manufacturing disruptions, regulatory compliance issues, and insufficient investment in production facilities. Many manufacturers operate within highly competitive markets where profit margins remain extremely limited. When a major facility experiences quality failures or regulatory shutdowns, alternative suppliers may not have enough available capacity to meet demand. This combination of operational and economic challenges frequently leads to prolonged supply shortages.
The proposed amendments would permit pharmaceutical alternatives containing different salts, esters, or complexes of the same active moiety to seek approval through the ANDS pathway. This change could improve patient access to lower-cost treatment options and increase market competition. However, concerns have been raised regarding potential differences in stability, impurity formation, safety profiles, and therapeutic performance. As a result, manufacturers may need to provide extensive supporting data to demonstrate equivalence.
Vaporized hydrogen peroxide (VHP) is widely used as an automated decontamination method within closed aseptic manufacturing systems. The process distributes sterilizing vapor throughout the isolator, effectively eliminating microorganisms from internal surfaces and equipment. Because the system remains sealed during decontamination, the risk of operator-related contamination is significantly reduced. VHP plays an important role in maintaining the sterile conditions required for aseptic fill-finish operations.
PLGA and PLA are biodegradable polymers commonly utilized in long-acting injectable formulations to provide controlled and sustained drug release. These materials encapsulate the active pharmaceutical ingredient within a polymer matrix that gradually breaks down after administration. As hydrolysis of the polymer occurs, the drug is released in a predictable manner over an extended period. This controlled-release mechanism helps maintain therapeutic drug levels while reducing dosing frequency.
Reference:
- U.S. Food and Drug Administration. (2026, May 26). Upcoming product-specific guidances for generic drug product development. U.S. Department of Health and Human Services. FDA: Upcoming Product-Specific Guidances for Generic Drug Product Development
- Health Canada. (2023, January 18). Access to generic drugs in Canada. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/fact-sheets/access-to-generic-drugs.html
- Tsakalozou, E., Gong, Y., Babiskin, A., Hu, M., Mousa, Y., Walenga, R., Wu, F., Yoon, M., Raney, S. G., Polli, J. E., Schwendeman, A., Krishnan, V., Fang, L., & Zhao, L. (2024). Application of advanced modeling approaches supporting generic product development under GDUFA for fiscal year 2023. AAPS Journal, 26(3), Article 72. https://doi.org/10.1208/s12248-024-00924-8
- U.S. Food and Drug Administration. (2024, November 7). Introduction and overview of the model master file (MMF) [Presentation slides]. U.S. Department of Health and Human Services. https://www.fda.gov/media/187989/download
- U.S. Food and Drug Administration. (2023). Population pharmacokinetic analyses — Format and content: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/166572/download
- European Medicines Agency (EMA). (2025). ICH Q3E extractables and leachables – Scientific guideline (Draft guideline for consultation). European Medicines Agency. https://www.ema.europa.eu/en/ich-q3e-extractables-leachables-scientific-guideline
- U.S. Pharmacopeia (USP). (2023). Addressing barriers to the development of complex generics: Understanding challenges and opportunities (Version 3.0). U.S. Pharmacopeia. https://www.usp.org/sites/default/files/usp/document/ea83b_complex-generics_wp_2023-07_v3.pdf
- Health Canada. (1990, September 24). Submissions for generic topical drugs. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/policies/submissions-general-topical-drugs.html

