
Introduction:
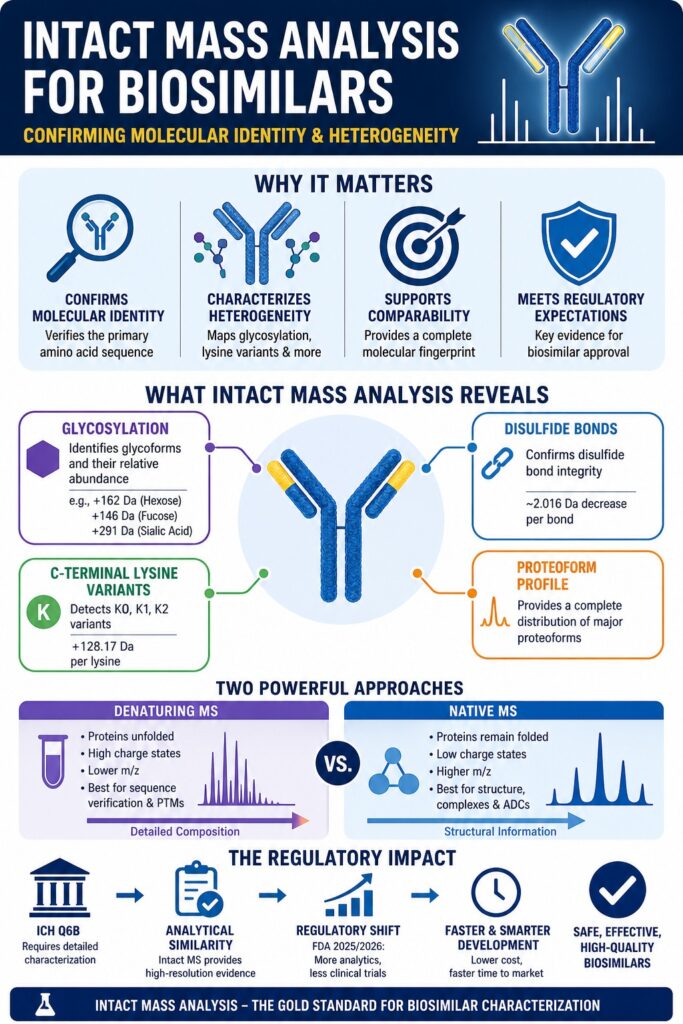
The accurate confirmation of molecular identity and the detailed characterization of heterogeneity are among the most essential analytical steps in the development of therapeutic proteins. Intact Mass Analysis Biosimilars is widely used as a high-resolution accurate-mass (HRAM) technique to confirm that the primary amino acid sequence of a biosimilar candidate matches that of its reference medicinal product (RMP). At the same time, it enables comprehensive profiling of post-translational modifications (PTMs). By analyzing the mass-to-charge ratio (m/z) of the intact, undigested protein, scientists obtain a complete molecular “fingerprint.” This fingerprint reflects the overall structural state, including glycosylation patterns, C-terminal processing, and truncations that may not be detected through bottom-up peptide mapping. In addition, this approach reduces the risk of overlooking subtle but functionally relevant variations that could affect therapeutic performance. It also supports early-stage comparability assessments, helping streamline development timelines and decision-making.
Explore specialized services: Biosimilar Characterization Using Mass Spectrometry
Share via:
The Strategic Importance of Intact Mass Analysis Biosimilars
Intact mass analysis biosimilars provides a fast and reliable way to verify the protein’s primary structure along with its major variants, without introducing artifacts that often arise during enzymatic digestion. This method plays a central role in the “totality of evidence” framework required by regulatory agencies, acting as an initial checkpoint to establish that a biosimilar is highly similar to its reference product. Unlike bottom-up proteomics, which fragments the protein into peptides, intact analysis preserves the molecule’s full structure, allowing detection of unexpected modifications such as internal cleavages, disulfide bond rearrangements, or incomplete signal peptide removal. This ensures that any deviations are observed within their true biological context. Additionally, the method improves confidence in comparability studies by providing a holistic view rather than fragmented insights.
Advances in biopharmaceutical manufacturing have shifted the focus of biosimilarity assessment from large clinical trials toward detailed analytical characterization. Modern mass spectrometry technologies can identify even minor structural differences with sensitivity far beyond that of clinical endpoints. As a result, generating a precise and reproducible intact mass profile has become a key factor in regulatory approval. This shift not only reduces development costs but also accelerates time to market. It also emphasizes the importance of analytical consistency across batches, which is critical for maintaining product quality throughout the lifecycle.
Our Analytical Capabilities: Analytical Services for Generic Drug Development
| Analytical Level | Data Output | Regulatory Relevance |
| Intact Level | Global MW, Glycan distribution, Lysine clipping | Identity, Heterogeneity, Consistency |
| Subunit Level | Domain-specific PTMs, Disulfide integrity | Regional quality attributes, Folding |
| Peptide Level | Sequence coverage, Trace PTM localization | Primary sequence, Site-specific modifications |
| Glycan Level | Linkage analysis, Sialylation, Monosaccharides | Biological activity, Immunogenicity |
Technological Foundations of High-Resolution Mass Spectrometry
The performance of Intact Mass Analysis Biosimilars depends heavily on the resolution and mass accuracy of the mass spectrometer, commonly using Orbitrap or Quadrupole Time-of-Flight (Q-TOF) systems. These instruments must resolve complex charge-state distributions of large proteins, such as monoclonal antibodies (~150 kDa), and differentiate between glycoforms that vary by only a few Daltons. High sensitivity and stability are essential to ensure reproducibility across multiple runs and samples. Furthermore, improvements in detector technology and ion optics have significantly enhanced signal clarity, even for low-abundance species. These capabilities are particularly important when analyzing complex biological matrices.
Orbitrap Mass Spectrometry in Intact Analysis
Orbitrap instruments trap ions within an electrostatic field, where they oscillate at frequencies proportional to the square root of their m/z ratio. This design enables extremely high resolution, reaching up to 1,000,000 FWHM at m/z 200, along with sub-1 ppm mass accuracy. Such precision makes Orbitrap systems ideal for detecting subtle micro-heterogeneity in biosimilars. They also maintain strong sensitivity across a broad mass range, which is advantageous for native mass spectrometry applications involving large protein complexes. In addition, their stability over long analytical runs supports consistent data generation. This makes them particularly valuable in regulated environments where reproducibility is critical.
Q-TOF Platforms and Fast Acquisition
Quadrupole Time-of-Flight (Q-TOF) systems offer a practical alternative, combining high acquisition speed with strong sensitivity. These features make them well suited for integration with ultra-high-performance liquid chromatography (UHPLC). Although earlier versions had lower resolution than Orbitrap systems, modern Q-TOF instruments have improved significantly. Innovations such as Electron Activated Dissociation (EAD) enhance fragmentation efficiency and enable better characterization of large proteins in complex samples like serum. Their rapid data acquisition also supports high-throughput workflows. This is especially beneficial in industrial settings where large numbers of samples must be analyzed efficiently.
Comparative Performance of HRMS Systems
| Feature | Orbitrap Technology | Q-TOF Technology |
| Primary Advantage | Highest resolving power & mass accuracy | High speed, sensitivity, & dynamic range |
| Typical Resolution | 140,000 – 1,000,000 | 40,000 – 80,000 |
| Mass Accuracy | < 1 ppm | 1 – 5 ppm |
| Ideal Application | Complex PTM profiling, Native MS | Rapid screening, LC-coupled analysis |
| Data Type | Fourier Transform based | Time-of-flight based |
Confirming Molecular Identity and Sequence Fidelity
The central objective of Intact Mass Analysis Biosimilars is to confirm that the protein backbone matches the intended amino acid sequence. This is achieved by comparing the experimentally measured mass with the theoretical mass derived from the sequence, including expected modifications such as disulfide bonds. This comparison provides a direct and reliable validation of molecular identity. It also serves as an important checkpoint before progressing to more detailed structural analyses.
Identity confirmation extends beyond simple molecular weight measurement. Even small deviations can indicate issues such as genetic mutations, amino acid substitutions, or incomplete signal peptide processing. Because intact mass analysis evaluates the entire molecule, it captures large-scale discrepancies that might be missed in peptide-level studies. This makes it an essential safeguard in quality control. It also helps detect rare variants that could impact efficacy or safety.
Support for Complex Molecules: Generic Peptide & Oligonucleotide Projects
Theoretical vs. Accurate Mass Calculation
In biosimilar analysis, “accurate mass” refers to the experimentally measured value, while “theoretical mass” is calculated from the atomic composition of the protein ($C, H, N, O, S$). For large proteins, analysts often use average mass values that account for natural isotopic abundance. This approach simplifies interpretation when individual isotopes cannot be resolved. It also improves consistency when comparing results across different instruments. Accurate mass calculations are essential for identifying small but meaningful differences between samples.
Verification of Disulfide Bond Integrity
Monoclonal antibodies typically contain 16 disulfide bonds that are critical for structural stability and biological function. Each bond formation results in a mass decrease of approximately 2.016 Da. Intact mass analysis under non-reducing conditions allows confirmation that all expected bonds are present. Any deviation from the expected mass may indicate incorrect folding or structural instability. This information is crucial for ensuring product quality and therapeutic reliability. It also provides insight into the robustness of the manufacturing process.
Mapping Molecular Heterogeneity and Major Proteoforms
Intact Mass Analysis Biosimilars is particularly effective for assessing macro-heterogeneity arising from natural variations in protein synthesis and processing. Biosimilars are not identical copies but consist of closely related proteoform distributions. Characterizing this distribution is essential for demonstrating similarity to the reference product. It also helps identify process-related variations that may need optimization. Understanding heterogeneity is key to maintaining consistent product performance.
Glycosylation Distribution and Relative Abundance
Glycosylation is a major contributor to heterogeneity in biotherapeutics, especially in IgG antibodies where N-linked glycans are attached at Asn297. Intact mass analysis reveals glycoform distributions as distinct peaks corresponding to specific sugar modifications. For example, each hexose addition increases mass by 162.14 Da, fucosylation adds 146.14 Da, and sialylation contributes 291.26 Da. These modifications can significantly influence biological activity, including immune response and serum half-life. Monitoring glycosylation patterns is therefore critical for ensuring consistent therapeutic performance. It also provides valuable information for process optimization.
C-terminal Lysine Variants and Bioactivity
Monoclonal antibodies often undergo C-terminal lysine clipping during production, resulting in K0, K1, and K2 variants. These differ by mass increments of 128.17 Da per lysine residue. Although these variants generally have minimal clinical impact, they are important indicators of process consistency. Intact mass analysis enables precise quantification of these forms. This ensures that the biosimilar matches the reference product’s heterogeneity profile. It also supports batch-to-batch consistency assessments.
Development Support: Formulation Development for Generic Drugs
| Proteoform Component | Component Mass (Da) | Cumulative Mass Effect |
| Protein Backbone | ~145,000 | Base Identity |
| G0F Glycan | 1,445 | Identity + Heterogeneity |
| G1F Glycan | 1,607 | +162 Da (Hexose) |
| G2F Glycan | 1,769 | +324 Da (2x Hexose) |
| C-terminal Lysine | 128 | +128 Da per Lysine |
Denaturing vs. Native Analytical Paradigms
Choosing between denaturing and native Intact Mass Analysis Biosimilars depends on the analytical objective. Denaturing methods focus on chemical composition, while native methods preserve higher-order structure. Using both approaches provides complementary data for a complete characterization. This combined strategy improves confidence in analytical results. It also supports more robust regulatory submissions.
Denaturing LC-MS: The Standard for Sequence Verification
Denaturing LC-MS uses reversed-phase chromatography with acidic conditions to unfold proteins. This exposes internal residues for protonation, resulting in high charge states and lower $m/z$ values. The large number of charge states improves deconvolution accuracy. This method is widely used for precise molecular weight determination and PTM analysis. It also offers high reproducibility, making it suitable for routine testing.
Native MS: Preserving Structure and Non-covalent Bonds
Native MS maintains proteins in their folded state using neutral buffers such as ammonium acetate. This results in lower charge states and higher $m/z$ values. It is especially useful for studying non-covalent complexes, bispecific antibodies, and antibody-drug conjugates. Native MS also provides better signal clarity for large assemblies. This makes it an important tool for advanced structural analysis.
- Maintains noncovalent interactions, which is critical for the study of bispecific antibodies and multimeric protein complexes.
- Enables intact analysis of antibody–drug conjugates (ADCs); unlike denaturing conditions that separate cysteine-linked ADCs into light and heavy chains, native mass spectrometry preserves the full structure for accurate drug-to-antibody ratio (DAR) measurement.
- Improves spectral quality, as folded proteins typically have smaller collisional cross-sections in the gas phase, reducing ion fragmentation and producing clearer spectra for large molecular assemblies.
Comparison of Mass Spectrometric Conditions
| Feature | Denaturing MS | Native MS |
| Chromatography | Reversed-Phase (RPLC) | Size-Exclusion (SEC) or IEX |
| pH Range | Acidic (pH ~2) | Neutral (pH ~7) |
| Charging Behavior | High z / Lower m/z | Low z / Higher m/z |
| Information Yield | Primary sequence & PTMs | Stoichiometry & HOS |
| Best For | Routine glycan profiling | Non-covalent complexes & ADCs |
Advanced Deconvolution and Informatics in Intact Analysis
The data generated from intact mass analysis requires complex deconvolution to produce a neutral mass spectrum. Modern algorithms such as UniDec and Bayesian-based approaches improve accuracy and reduce artifacts. These tools enable better interpretation of complex spectra. They also support automated workflows, reducing manual intervention. Advances in informatics continue to enhance data reliability and throughput.
The Evolution from MaxEnt to Modern Bayesian Algorithms
For many years, the Maximum Entropy (MaxEnt1) approach has served as a standard method for spectral deconvolution. It relies on an objective function that balances goodness of fit and peak sharpness while maximizing entropy to distinguish closely spaced mass signals. Despite its effectiveness, this method can sometimes overfit the data, producing excess peaks and generating so-called harmonic artifacts—spurious signals that appear at fractional or multiple values of the true mass.
More recent computational approaches have been developed to overcome these challenges:
- UniDec: A Bayesian-driven method that removes the entropy constraint and instead applies smoothing across adjacent charge states, making it well suited for heterogeneous samples and ion mobility datasets.
- BayesSpray: A nested sampling–based technique that automatically optimizes its parameters, enabling reliable interpretation of both well-resolved and complex, unresolved spectra.
- Parsimonious Charge Deconvolution: This strategy uses a two-step workflow—first assigning charge states, then refining peak shapes—to identify the smallest set of masses needed to explain the observed data, thereby minimizing harmonic and side-lobe artifacts.
Managing Informatics Hurdles
Processing intact protein data requires advanced informatics tools capable of performing techniques such as sliding window deconvolution, where spectra are averaged across an elution profile to improve sensitivity and mass accuracy. This capability is especially important for detecting low-abundance modifications, such as lysine glycation (+162.1 Da) or minor oxidative changes (+15.99 Da), which may otherwise be obscured by baseline noise in individual scans.
Subunit and Middle-Up Analysis Strategies
Subunit analysis breaks large proteins into smaller fragments, improving resolution and sensitivity. Techniques like IdeS digestion produce fragments that are easier to analyze. This approach bridges the gap between intact and peptide-level analysis. It also allows detection of subtle modifications with higher confidence. As a result, it is increasingly used in detailed characterization studies.
The Role of IdeS (FabRICATOR) Digestion
IdeS (FabRICATOR), an enzyme originating from Streptococcus pyogenes, selectively cleaves IgG antibodies just below the hinge region. When this enzymatic digestion is followed by reduction, the antibody is split into three fragments of approximately 25 kDa each: the light chain (LC), the Fc/2 segment, and the Fd′ fragment. Analyzing these smaller units improves signal-to-noise ratios and allows for more confident detection of subtle mass differences compared to examining the intact ~150 kDa molecule.
Multi-Attribute Method (MAM) Integration
Intact and subunit analysis are increasingly being integrated into Multi-Attribute Methods (MAM). By replacing multiple traditional assays (e.g., CEX for charge variants, HILIC for glycans) with a single, high-resolution LC-MS run at the subunit level, manufacturers can achieve faster turnaround times and more comprehensive data on critical quality attributes. This approach is highly favored by regulatory agencies because it provides direct, quantitative information on the molecular level.
Specialized Product Services: Semaglutide Generic Projects
| Liraglutide Development Services
| Lanreotide Development Services
| Fragment Type | Typical Mass (kDa) | Analytical Insight |
| Intact mAb | ~150 | Global fingerprint, identity |
| F(ab’)2 | ~100 | Antigen binding integrity |
| Fc fragment | ~50 | Effector function, FcRn binding |
| LC / HC / Fd’ | ~25 | High-res glycan & PTM profiling |
Regulatory Landscape: ICH Q6B and the FDA 2025/2026 Shift
Regulatory guidelines such as ICH Q6B require detailed characterization of biotherapeutics, including molecular weight and structural variants. Recent FDA and EMA updates emphasize analytical data over large clinical trials. This reflects growing confidence in advanced analytical technologies. The shift reduces development costs and speeds up approval timelines. It also highlights the importance of high-quality analytical methods.
ICH Q6B: The Foundation of Characterization
ICH Q6B requires determination of molecular weight and overall molecular characteristics using suitable analytical techniques, with mass spectrometry widely recognized as a key method for confirming primary structure and identifying isoforms. The guideline also highlights the need to evaluate glycan structures and to detect product-related variants, including truncated species or improperly processed forms.
The 2025/2026 Regulatory Pivot
Recent updates in regulatory thinking indicate a significant shift in approach. Draft guidances released by the FDA in late 2025 and early 2026 place strong emphasis on analytical comparability studies along with focused pharmacokinetic (PK) evaluations, while reducing reliance on large-scale comparative efficacy trials (CES). The underlying rationale is that when a biosimilar demonstrates a highly similar “fingerprint” to the reference product using advanced tools such as high-resolution mass spectrometry, the remaining uncertainty is minimal, potentially eliminating the need for Phase III trials. This shift can substantially lower development costs and shorten timelines to market entry.
Demonstrating Analytical Similarity
To meet regulatory expectations, developers must show that the characteristics of the biosimilar fall within the natural variability observed across multiple batches of the reference product. This involves analyzing several lots of both the biosimilar and the innovator product to establish statistical comparability. Intact mass analysis plays a central role in this tiered evaluation strategy, delivering the quantitative evidence needed to confirm that variations in critical quality attributes, such as glycosylation patterns, do not impact clinical performance.
Ensuring Compliance: Regulatory Support for Generic Drug Development
Method Validation and Sample Preparation Considerations
Accurate results depend on careful sample preparation to avoid artificial modifications. Factors such as oxidation, deamidation, and disulfide scrambling must be minimized. Simple preparation methods are preferred to preserve the protein’s native state. Proper validation ensures reliable and reproducible results. This is essential for meeting regulatory standards.
Minimizing Artificial Modifications
Sample preparation approaches used for small molecules are not suitable for large biomolecules such as proteins. Excessive handling can introduce unintended changes, including:
- Artificial Oxidation: Triggered by exposure to light, elevated temperatures, or contact with metal surfaces.
- Deamidation: Promoted by extreme pH conditions during sample processing.
- Disulfide Scrambling: Resulting from incorrect reduction or denaturation procedures.
To prevent these artifacts, intact mass analysis emphasizes minimal sample manipulation, often limited to simple dilution in a suitable spray solvent such as ammonium acetate or water. When buffer exchange is required, centrifugal filtration using ammonium bicarbonate is commonly employed to eliminate sodium adducts, which can otherwise lead to peak broadening and diminished spectral resolution.
Scale-up Solutions: Manufacturing Scale-up for Generic Drugs
Comparative Data Analysis: Mirror Plots and Statistical Equivalence
Mirror plots provide a visual comparison between biosimilars and reference products, highlighting similarities in mass and intensity. Quantitative analysis of peak intensities supports statistical equivalence testing. Together, these methods demonstrate analytical similarity. They also provide clear evidence for regulatory review. Effective data presentation is key to successful submissions.
Mirror Plots for Visual Confirmation
Mirror plots provide an effective way to visually compare datasets by displaying the deconvoluted spectrum of the reference medicinal product (RMP) above the x-axis and that of the biosimilar candidate below it. This format enables rapid assessment of whether key features—such as glycoforms, lysine variants, and any detectable impurities—correspond in both mass and relative intensity. When the two spectra closely resemble one another, the resulting “fingerprint-like” pattern offers strong visual support for analytical similarity.
Quantifying Relative Abundance of Variants
In addition to qualitative comparison, it is essential to quantify the relative distribution of each proteoform. This is typically achieved by measuring peak intensities or calculating the area under the curve (AUC) for each deconvoluted mass signal. The resulting data are then subjected to statistical equivalence evaluations to confirm that the biosimilar falls within established acceptance criteria, which are defined based on the variability observed across multiple batches of the reference product throughout its production history.
Conclusions and Future Outlook
Intact Mass Analysis Biosimilars remains a cornerstone technique for confirming molecular identity and heterogeneity in biotherapeutics. It delivers a detailed and comprehensive view of protein structure and modifications. Ongoing advancements in mass spectrometry and data analysis continue to improve accuracy and efficiency. As regulatory frameworks evolve, the importance of analytical characterization will only increase. Mastering these techniques is essential for developing safe, effective, and high-quality biosimilars.
Frequently Asked Questions (FAQs)
Intact mass analysis checks the full molecular weight of the protein and compares it with a calculated value based on its amino acid sequence. When both values closely match within a very small margin, it confirms that the sequence is correct. This also verifies proper protein expression and correct disulfide bond formation during production.
Common mass changes are mainly linked to glycosylation and terminal modifications. For example, adding sugar units increases the mass in predictable steps, while C-terminal lysine adds a defined increment. Other small changes, such as oxidation or deamidation, may also occur but are usually easier to confirm in more detailed analyses.
Denaturing mass spectrometry unfolds the protein using acidic conditions, allowing accurate measurement of its core structure and molecular weight. In contrast, native mass spectrometry keeps the protein in its natural folded state. This helps preserve weak interactions and provides insight into protein complexes and functional forms.
Harmonic artifacts are incorrect signals that can appear during data processing when charge states are assigned wrongly. These false peaks may look like real impurities or structural variants. If not identified properly, they can lead to incorrect conclusions about the sample’s quality.
ICH Q6B guidelines require detailed evaluation of protein structure, including molecular weight and product consistency. Intact mass analysis supports these requirements by providing clear data on protein identity and variation. It is widely accepted as a standard method to meet these regulatory expectations.
Middle-up analysis involves breaking a large protein into smaller sections before mass analysis. This approach improves detection of small modifications that may not be visible in full protein analysis. It is especially useful when more detailed information about specific regions of the protein is needed.
The FDA reviews all available data, including analytical, non-clinical, and clinical results, to assess biosimilarity. Analytical similarity forms the foundation of this approach. Strong analytical data, such as from intact mass analysis, can reduce the need for extensive clinical studies.
C-terminal lysine clipping is a common modification where lysine residues are removed from the protein end. Although it usually does not affect the drug’s function, it is important for consistency. Matching this pattern with the reference product helps confirm similar manufacturing quality.
Proteins can easily change if handled improperly during preparation. Conditions like extreme pH or exposure to air can introduce unwanted modifications. Careful and simple preparation ensures that the results reflect the true structure of the protein rather than lab-induced changes.
Reference:
- Yang, Y., Wang, G., Song, T., Lebrilla, C. B., & Heck, A. J. R. (2017). Resolving the micro-heterogeneity and structural integrity of monoclonal antibodies by hybrid mass spectrometric approaches. mAbs, 9(4), 638–645. https://doi.org/10.1080/19420862.2017.1295207
- Heinemann, L., & Hompesch, M. (2018). Biosimilar insulins: Basic considerations. Journal of Diabetes Science and Technology, 12(1), 4–9. https://doi.org/10.1177/1932296817745939
- Kafader, J. O., Melani, R. D., Schachner, L. F., Ives, A. N., Patrie, S. M., Kelleher, N. L., & Compton, P. D. (2020). Native vs denatured: An in-depth investigation of charge state and isotope distributions. Journal of the American Society for Mass Spectrometry, 31(3), 574–581. https://doi.org/10.1021/jasms.9b00040
- European Medicines Agency. (1999). ICH Q6B: Specifications: Test procedures and acceptance criteria for biotechnological/biological products (CPMP/ICH/365/96). https://www.ema.europa.eu/en/ich-q6b-specifications-test-procedures-acceptance-criteria-biotechnological-biological-products-scientific-guideline
- Bern, M., Caval, T., Kil, Y. J., Tang, W., Becker, C., Carlson, E., Kletter, D., Sen, K. I., Galy, N., Hagemans, D., Franc, V., & Heck, A. J. R. (2018). Parsimonious charge deconvolution for native mass spectrometry. Journal of Proteome Research, 17(3), 1216–1226. https://doi.org/10.1021/acs.jproteome.7b00839
- Chow, S.-C., Song, F., & Bai, H. (2018). Analytical similarity assessment in biosimilar product development. The AAPS Journal, 18(3), 670–677. https://doi.org/10.1208/s12248-016-9882-5

