
Introduction
Developing a generic version of leuprolide acetate depot injections is considered one of the most challenging tasks in the complex generics field. Unlike conventional oral generics, the Leuprolide Depot ANDA Requirements involve advanced formulation development, detailed polymer characterization, and evaluation of long-acting drug release behavior. These formulations must replicate both the active pharmaceutical ingredient and the specialized drug delivery system that controls its release over time.
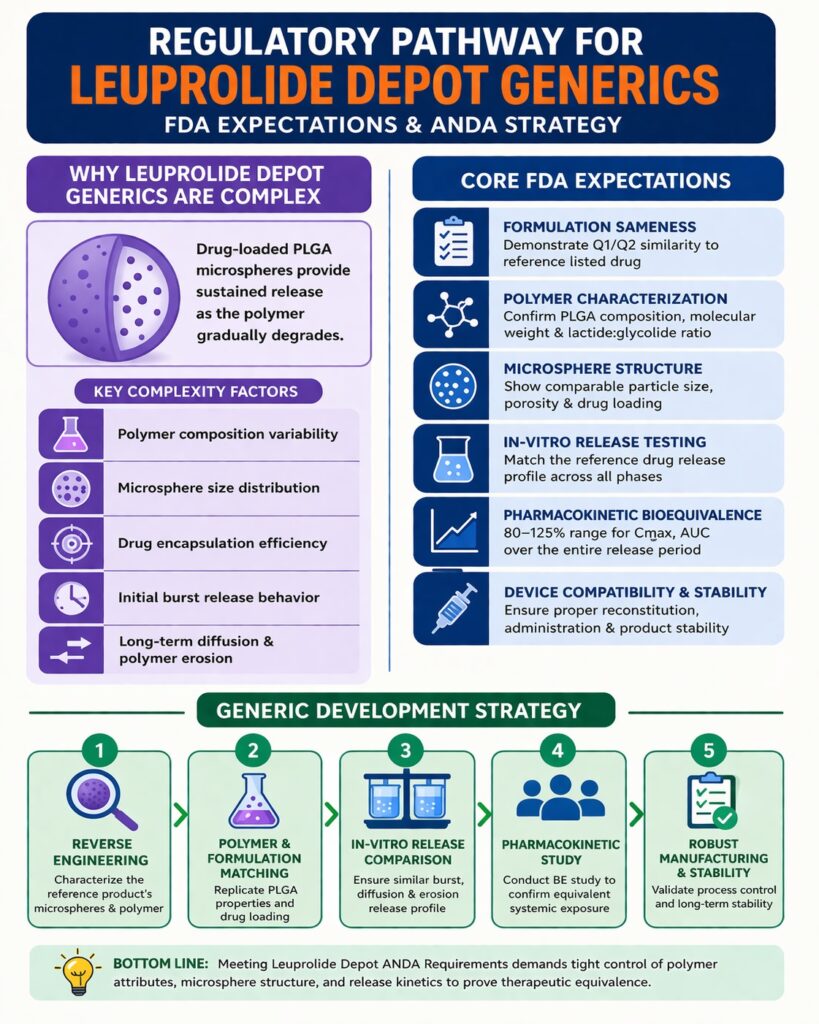
Leuprolide depot products, such as the reference listed drug Lupron Depot®, use poly(lactic-co-glycolic acid) (PLGA) microspheres to provide sustained drug release. These biodegradable microspheres gradually degrade after injection and slowly release the peptide drug into systemic circulation. Because the formulation combines drug, polymer, and delivery mechanism, it functions as a complex therapeutic system rather than a simple injectable solution.
As a result, regulatory agencies classify these formulations as complex parenteral drug products. The Leuprolide Depot ANDA Requirements therefore demand extensive analytical characterization, formulation similarity, and pharmacokinetic bioequivalence data. This article explains the FDA expectations and regulatory strategies that companies must follow when developing generic leuprolide depot injections.
Explore our specialized expertise in this field: Learn more about our Leuprolide Depot Generic Drug Development Services
Share via:
Why Leuprolide Depot Generics Are Considered Complex
Leuprolide depot injections are considered complex drug products because they rely on biodegradable polymer microspheres to control drug release over extended periods. These microspheres act like tiny reservoirs that hold the drug inside the polymer matrix. After injection, the polymer slowly degrades and releases the active drug into the body in a controlled way.
The microspheres contain leuprolide embedded within PLGA polymer matrices. Over time, the polymer undergoes hydrolysis, which breaks the polymer chains into smaller components such as lactic acid and glycolic acid. As the matrix breaks down, the drug gradually diffuses out of the microspheres and enters systemic circulation.
Several formulation factors strongly influence how these depot systems behave. These include the polymer composition, particle size distribution, and drug loading efficiency within the microspheres. Even small changes in these parameters can significantly affect the drug release pattern and therapeutic performance of the product.
Key complexities include:
| Major Complexity Factors |
|---|
| Polymer composition variability |
| Microsphere size distribution |
| Drug encapsulation efficiency |
| Initial burst release behavior |
| Long-term diffusion and polymer erosion mechanisms |
Each of these parameters affects the overall pharmacokinetic profile of the injection. If any attribute differs too much from the reference product, the drug exposure in patients may change. This could influence both the safety and effectiveness of the therapy.
Because of this sensitivity, developers must carefully control every stage of formulation development and manufacturing. Even minor variations in polymer molecular weight or microsphere porosity can change the drug release rate. Therefore, detailed analytical testing and tight process control are essential when meeting Leuprolide Depot ANDA Requirements.
Scientific studies show that PLGA-based long-acting injectables require thorough characterization because their performance depends heavily on polymer molecular weight, lactide-to-glycolide ratio, and particle morphology. Accurate measurement of these attributes helps confirm that the generic formulation behaves similarly to the reference product.
Understand the technical hurdles involved: Read about Leuprolide Depot Analytical Challenges
Core FDA Expectations for Leuprolide Depot ANDA Requirements
The FDA expects a comprehensive scientific evidence package for approval of leuprolide depot generics. Because these formulations involve complex delivery systems, regulators evaluate several aspects of the product simultaneously. Analytical data, manufacturing controls, and clinical performance studies must all support the claim of equivalence.
The main regulatory pillars within the Leuprolide Depot ANDA Requirements include formulation sameness, polymer characterization, microsphere structure analysis, in-vitro release testing, pharmacokinetic bioequivalence, device compatibility, and product stability studies.
Regulatory expectations include demonstrating Q1/Q2 similarity with the reference product, confirming PLGA polymer composition, and establishing comparable microsphere morphology and drug loading levels. Developers must also show that the in-vitro drug release profile closely matches that of the reference formulation.
Review the foundational standards for generic submissions: Requirements for ANDA Submission of Generic Drugs
Bioequivalence studies must evaluate pharmacokinetic exposure across the entire drug release cycle, which may last several weeks or months. These studies measure systemic drug levels over time to confirm that the generic product releases the drug at the same rate and extent as the original product.
Device compatibility is another important requirement. Many depot injections use specialized devices for reconstitution and administration. The drug formulation must work smoothly with the delivery system to ensure accurate dosing and consistent drug suspension.
Stability studies must also demonstrate that both the polymer matrix and peptide drug remain stable throughout the product’s shelf life. Changes in polymer degradation or drug potency during storage could affect the release profile. Therefore, regulators require long-term stability data as part of the ANDA submission.
Together, these elements form the core scientific framework of the Leuprolide Depot ANDA Requirements, ensuring that the generic product performs the same as the reference drug in patients.
Reverse Engineering Strategy for Generic Development
A strong generic development strategy often begins with reverse engineering of the reference listed drug. This process involves analyzing the physical and chemical properties of the original product to understand how the formulation works. By studying the reference formulation carefully, developers can identify the critical parameters that control drug release.
Reverse engineering requires detailed analytical characterization of the reference product. Scientists use techniques such as microscopy, chromatography, and thermal analysis to examine the microsphere structure and polymer properties. These methods provide important insights into particle morphology, polymer composition, and drug distribution within the microspheres.
Follow the roadmap for success: How to Develop Generic Leuprolide Depot
Important analytical parameters include polymer molecular weight distribution, residual solvent levels, microsphere porosity, and drug encapsulation efficiency. Particle size distribution and surface structure are also evaluated because they influence drug diffusion and polymer degradation.
Additional characterization may include evaluation of polymer end groups, crystallinity, and thermal behavior. These factors influence how the polymer interacts with the drug and how it degrades in the body. Analytical techniques such as differential scanning calorimetry and gel permeation chromatography are commonly used for this purpose.
Research consistently shows that matching PLGA polymer composition and microsphere structure is essential for reproducing the release behavior of the reference product. If polymer characteristics differ significantly, the drug release profile may change, which can affect therapeutic outcomes.
Therefore, reverse engineering plays a major role in meeting Leuprolide Depot ANDA Requirements. Accurate characterization of the reference product helps developers design a generic formulation with similar structural and functional properties.
Polymer Characterization and PLGA Equivalence in Leuprolide Depot ANDA Requirements
One of the most carefully evaluated aspects of the Leuprolide Depot ANDA Requirements is polymer characterization. PLGA polymers form the structural framework that controls drug release in depot formulations. Their chemical structure and molecular weight determine how quickly the polymer degrades in the body.
The ratio between lactic acid and glycolic acid units strongly influences polymer degradation behavior. Polymers with higher lactide content tend to degrade more slowly, while those with higher glycolide content break down faster. Therefore, matching the lactide:glycolide ratio of the reference product is critical for achieving similar drug release patterns.
Regulators evaluate several important polymer attributes during the review process. These include molecular weight distribution, polydispersity index, end-group chemistry, residual monomer content, and glass transition temperature. Each of these parameters provides insight into the structure and stability of the polymer.
Get support from a specialized partner: Choosing the right CRO for Leuprolide Depot Development
Common analytical methods used for polymer characterization include gel permeation chromatography (GPC), nuclear magnetic resonance (NMR) spectroscopy, and differential scanning calorimetry (DSC). These techniques help determine whether the polymer used in the generic formulation closely matches that of the reference product.
Scientific research shows that even small differences in PLGA properties can affect drug release kinetics. For example, slight changes in molecular weight or end-group chemistry can alter polymer degradation rates. These variations may ultimately influence pharmacokinetic performance.
For this reason, developers must provide strong analytical evidence demonstrating polymer equivalence. Accurate polymer selection and detailed characterization are essential steps in satisfying the Leuprolide Depot ANDA Requirements.
In-Vitro Release Testing and IVIVC Considerations
FDA reviewers place significant importance on comparative in-vitro release testing for depot microsphere formulations. These laboratory studies evaluate how the drug is released from the polymer matrix over time under controlled conditions. The goal is to determine whether the generic formulation produces a release profile similar to the reference product.
PLGA microspheres typically show a three-phase release pattern. The first phase is the initial burst release, where drug near the surface of the microspheres diffuses quickly into surrounding fluids. This phase occurs soon after injection and contributes to early systemic drug exposure.
The second phase is the diffusion-controlled stage, where drug molecules slowly migrate through the polymer matrix. This phase provides sustained drug release over an extended period.
The final phase occurs when the polymer begins to degrade significantly. As the matrix erodes, the remaining drug is released from the microspheres. Replicating these phases is important for meeting the Leuprolide Depot ANDA Requirements.
Analytical approaches may include specialized dissolution systems designed for long-acting injectables. Accelerated release tests and mathematical modeling may also be used to study release kinetics.
Developers often try to establish an in-vitro–in-vivo correlation (IVIVC) that links laboratory release data with pharmacokinetic results from clinical studies. Although creating strong IVIVC models for PLGA systems can be challenging, they can provide useful insights into product performance.
Learn more about analytical benchmarks: Analytical Requirements for ANDA Generic Drugs
Bioequivalence Study Design for Leuprolide Depot
One of the most important clinical components of the Leuprolide Depot ANDA Requirements is demonstrating pharmacokinetic bioequivalence. These studies confirm that the generic formulation delivers drug exposure comparable to the reference product.
Bioequivalence studies typically measure parameters such as Cmax, AUC0-t, and AUC0-∞. These values describe the peak concentration of the drug and the total systemic exposure over time.
Regulatory guidelines generally require the confidence intervals for these parameters to fall within 80–125% of the reference product values. Meeting this range confirms that the generic and reference products behave similarly in the body.
Because depot injections release drug slowly, clinical studies may require long sampling periods lasting several weeks or months. Researchers must collect blood samples at multiple time points to capture the entire drug release cycle.
Parallel-group study designs are commonly used since crossover designs may not be practical for long-acting injections. These studies often involve large participant groups and careful statistical analysis.
Although these studies are complex and costly, they play a crucial role in meeting Leuprolide Depot ANDA Requirements and confirming therapeutic equivalence.
Manufacturing and Process Control Expectations
Manufacturing processes for depot microspheres require precise control to ensure consistent product quality. These formulations are commonly produced using techniques such as solvent evaporation or phase separation.
The process usually begins with dissolving the polymer and drug in an organic solvent. This solution is then emulsified in an aqueous phase, and the solvent gradually evaporates. As the solvent disappears, the polymer forms solid microspheres that contain the drug.
Key process variables include solvent type, mixing speed, temperature control, evaporation rate, and drying methods. Each of these factors influences the final particle size, drug distribution, and microsphere structure.
Even small changes in manufacturing conditions can affect release kinetics. For this reason, FDA reviewers expect strong process validation and Quality-by-Design (QbD) strategies. These frameworks identify critical process parameters and establish acceptable operating ranges.
Consistent manufacturing control helps ensure that the product continues to meet the Leuprolide Depot ANDA Requirements throughout commercial production.
Accelerate your timeline with a CDMO: How a CDMO can accelerate generic drug development in the US and Canada
Device and Combination Product Considerations
Many leuprolide depot formulations are marketed as drug-device combination products. These systems often include specialized devices used to reconstitute and administer the microsphere suspension.
Examples include dual-chamber syringes or mixing systems that combine the drug and diluent immediately before injection. The device ensures proper suspension of the microspheres and accurate dosing.
For generic versions, the device must demonstrate functional equivalence with the reference product. Testing may evaluate mixing efficiency, injection force, and dose accuracy.
Usability studies are also conducted to confirm that healthcare professionals can operate the device safely and easily. These studies help ensure proper administration and reduce the risk of dosing errors.
Compatibility between the formulation and the device must also be demonstrated. The suspension should remain stable during mixing and injection without clogging or settling.
Meeting these criteria is another important component of the Leuprolide Depot ANDA Requirements.
Strategic FDA Interaction for Leuprolide Depot ANDA Requirements
Due to the complexity of depot formulations, early interaction with the FDA is strongly recommended. Regulatory meetings allow developers to discuss their plans and receive feedback from agency reviewers.
Pre-ANDA meetings are commonly used to present the overall development strategy. Companies may discuss reverse engineering data, polymer characterization plans, and proposed in-vitro release testing methods.
Product development meetings focus on technical issues related to formulation design and bioequivalence studies. Developers may present preliminary analytical results and receive regulatory guidance.
Pre-submission meetings sometimes occur shortly before filing the ANDA. These discussions help confirm that all required data are complete and aligned with regulatory expectations.
Such interactions help ensure that the development program meets the Leuprolide Depot ANDA Requirements and reduce the risk of regulatory delays.
Compare your outsourcing options: CRO vs. In-House ANDA Development
Risk Mitigation Strategies for ANDA Approval
Companies developing leuprolide depot generics often implement several strategies to reduce development risks. One key step is early and detailed characterization of the reference listed drug.
Developing predictive in-vitro release models can also help scientists evaluate formulation changes before conducting expensive clinical trials. These models allow developers to optimize the formulation more efficiently.
Using multiple analytical techniques provides a deeper understanding of polymer properties and microsphere structure. Combining methods such as microscopy, chromatography, and spectroscopy strengthens the scientific evidence for equivalence.
Implementation of Quality-by-Design (QbD) frameworks also improves manufacturing consistency and regulatory confidence. These approaches identify critical parameters and establish effective process controls.
Conducting small pilot pharmacokinetic studies can provide early insight into clinical performance. These studies help determine whether the formulation is likely to meet the Leuprolide Depot ANDA Requirements before large trials begin.
Ensure your product meets safety standards: Nitrosamine Risk Assessment in Generic Drugs
Conclusion
Developing generic versions of leuprolide depot injections requires a deep understanding of complex injectable drug systems and regulatory expectations. These products combine advanced polymer science with sophisticated drug delivery technology, making them far more challenging than conventional generics.
The Leuprolide Depot ANDA Requirements involve extensive analytical characterization, polymer equivalence studies, in-vitro release testing, and long-term pharmacokinetic evaluations. Each component provides critical evidence that the generic product performs the same as the reference formulation.
A successful development strategy begins with reverse engineering of the reference product, followed by robust formulation optimization and analytical validation. Developers must also establish reliable manufacturing processes that consistently produce microspheres with the desired properties.
Early communication with the FDA and strong CMC documentation greatly improve the chances of regulatory approval. By aligning development programs with current regulatory guidance, companies can reduce delays and improve submission success.
For organizations working on complex injectable generics, understanding and addressing the Leuprolide Depot ANDA Requirements is essential for successfully bringing these important therapies to market.
If you require technical or regulatory guidance on complex injectable generics or depot formulation characterization, contact our experts here: Contact us
Frequently Asked Questions (FAQs)
The main challenges involve matching the microsphere structure, polymer characteristics, and long-term drug release behavior of the reference product. Small differences in polymer composition or particle structure can change how the drug is released in the body. Therefore, developers must generate detailed analytical and clinical data to meet the Leuprolide Depot ANDA Requirements.
PLGA microspheres act as the controlled drug delivery system in these formulations. Their chemical composition and molecular weight determine how quickly the polymer degrades after injection. Because this process controls the timing and duration of drug release, regulators require detailed polymer characterization.
Several analytical methods are used to study microsphere formulations. Scanning electron microscopy helps visualize particle structure, while particle size analysis measures distribution patterns. Techniques such as DSC, GPC, and HPLC are used to evaluate polymer properties and drug loading efficiency.
In-vitro release testing is very important because it helps compare the release behavior of the generic product with the reference drug. These tests evaluate the burst release and sustained release phases of the formulation. Consistent results help predict similar pharmacokinetic performance.
IVIVC links laboratory release data with clinical pharmacokinetic results. When established successfully, it helps predict how the drug will behave in patients based on in-vitro tests. Although difficult to develop for PLGA systems, IVIVC models can strengthen regulatory submissions.
Reverse engineering allows developers to analyze the composition and structure of the reference product. By understanding polymer properties, drug loading levels, and microsphere morphology, scientists can design a generic product that behaves similarly. This step greatly supports compliance with Leuprolide Depot ANDA Requirements.
The FDA reviews polymer molecular weight distribution, monomer ratios, end-group chemistry, and degradation behavior. Analytical methods such as chromatography and spectroscopy are used to measure these attributes. Developers must demonstrate that their polymer closely matches the one used in the reference product.
Manufacturing microspheres requires precise control of processes such as emulsification, solvent evaporation, and polymer precipitation. Small changes in mixing conditions or temperature can alter particle size and drug distribution. These variations can affect drug release kinetics and overall product performance.
Reference:
- Chwalisz, K. (2023). Clinical development of the GnRH agonist leuprolide acetate depot. F&S Reports, 4(2 Suppl), 33–39. https://pmc.ncbi.nlm.nih.gov/articles/PMC10201295/
- Zhou, Z., Zhou, Y., Yan, W., Feng, T., & Liang, Z. (2024). Comparison of the efficacy and safety profiles of generic and branded leuprorelin acetate microspheres in patients with prostate cancer. Oncology Letters, 28(1), 319. https://pmc.ncbi.nlm.nih.gov/articles/PMC11130615/
- U.S. Food and Drug Administration. (2018). NDA 205054: Multi-disciplinary review and evaluation — Lutrate Depot (leuprolide acetate for depot suspension). Center for Drug Evaluation and Research. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/205054Orig1s000MultidisciplineR.pdf
- Swayzer, D. V., & Gerriets, V. (2023). Leuprolide. In StatPearls. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK551662/

