
Introduction: Why Conventional Analytical Methods Are Insufficient for Nitrosamines
For pharmaceutical manufacturers facing increasing regulatory scrutiny regarding genotoxic impurities, conventional compendial analytical methods are rarely adequate. Nitrosamine Method Development and Validation Services demand a fundamentally different analytical strategy. This approach begins by understanding acceptable intake (AI) limits, then working backward to establish the required sensitivity before designing an analytical system capable of consistently detecting and quantifying trace-level carcinogenic impurities within complex pharmaceutical matrices.
Following the valsartan-related nitrosamine contamination crisis of 2018–2019, regulatory agencies including the FDA, EMA, Health Canada, and PMDA implemented progressively more detailed requirements governing the development, characterization, and validation of nitrosamine analytical methods. These methods must be thoroughly validated before their data can support regulatory submissions, release testing programs, or stability studies. This article examines the key technical and regulatory considerations involved in developing scientifically sound and submission-ready nitrosamine analytical methods.
Learn more about our comprehensive Nitrosamine Analysis and Testing Services
Article Summary:
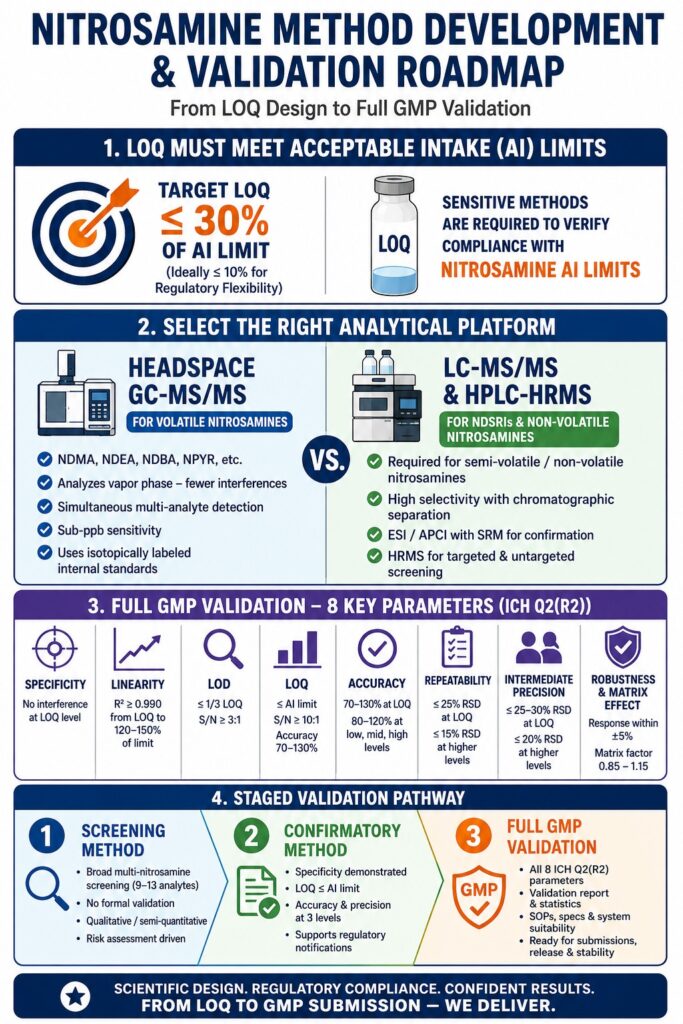
- Nitrosamine testing methods should be capable of quantifying impurities at concentrations that align with compound-specific Acceptable Intake (AI) requirements. To maintain regulatory compliance, laboratories commonly establish LOQs significantly below the permitted limit, often targeting 30% or less of the AI value.
- The selection of an analytical platform depends on several factors, including the volatility of the nitrosamine, the complexity of the sample matrix, and the chemical characteristics of the target impurity. Volatile nitrosamines are typically analyzed using headspace GC-MS/MS, while NDSRIs and other non-volatile compounds generally require LC-MS/MS or high-resolution mass spectrometry techniques.
- A complete GMP validation program in accordance with ICH Q2(R2) extends far beyond basic system suitability testing. The method must demonstrate acceptable performance for parameters such as specificity, linearity, detection and quantitation limits, recovery, precision, robustness, and matrix-effect control before it can be considered suitable for routine GMP use.
- Implementing a phased development strategy allows organizations to build analytical confidence while managing resources effectively. The process typically progresses from risk assessment and preliminary screening through confirmatory analysis before advancing to comprehensive GMP validation.
- Isotopically labeled internal standards, including compounds such as d₆-NDMA, d₁₀-NDEA, and d₁₄-NDBA, play a critical role in method accuracy. These standards help compensate for variations caused by extraction efficiency, instrument response, and matrix-related ion suppression or enhancement.
- Many nitrosamine method validation challenges can be traced back to early development decisions. Common issues include insufficient method sensitivity, incomplete matrix-effect characterization, inadequate intermediate precision studies, and the use of internal standards that do not appropriately match the target NDSRI.
What LOQ Actually Means in a Nitrosamine Context — and Why It Cannot Be Compromised
The Limit of Quantitation (LOQ) for a nitrosamine method must be established at or below the regulatory reporting threshold applicable to the specific nitrosamine under evaluation. In most cases, this means an LOQ at or below 30% of the acceptable intake limit, while many laboratories aim for ≤10% of the AI limit to provide additional regulatory flexibility.
This requirement is not optional. The FDA’s revised September 2024 guidance, Control of Nitrosamine Impurities in Human Drugs, explicitly states that sensitive analytical procedures with appropriate LOQs are necessary to verify compliance with recommended nitrosamine AI limits. Regulatory agencies also require that the chosen LOQ be scientifically justified, a requirement that reviewers generally evaluate with considerable rigor.
Explore Analytical Sensitivity Targets: For a deeper dive into establishing trace-level compliance margins, read our technical breakdown on achieving an Ultra-Low Limit of Quantitation (LOQ) in Nitrosamine Testing.
Practical LOQ Targets by Nitrosamine Class
| Nitrosamine | AI Limit (ng/day) | Target LOQ (ng/g or ppm) for an 880 mg MDD Product | Notes |
|---|---|---|---|
| NDMA | 96 | ≤0.03 ppm | FDA example limit from guidance |
| NDEA | 26.5 | ~0.01 ppm | Higher carcinogenic potency |
| NMBA | 96 | ≤0.03 ppm | Frequently relevant in sartan APIs |
| NPYR | 96 | ≤0.03 ppm | Suitable for GC-MS/MS headspace analysis |
| NDBA | 26.5 | ~0.01 ppm | Requires careful internal standard selection |
| NDSRIs (case-specific) | Determined through CPCA or in vivo/in vitro data | Structure-dependent | No universal limit; AI established according to ICH M7(R2) Appendix 1 |
The method LOQ must be reassessed whenever formulation composition, drug loading, or the manufacturing process changes. An LOQ validated for one pharmaceutical matrix cannot automatically be applied to another. Matrix-specific recovery studies are necessary to confirm continued suitability.
Evaluating High-Risk Formulations? Certain compound families require specialized screening considerations. Discover our approach to Nitrosamine Testing for High-Risk Drug Classes.
Analytical Platform Selection for Nitrosamine Method Development
Selecting the appropriate analytical platform is one of the most important decisions in nitrosamine method development. An unsuitable platform can generate inconsistent or unreliable data, regardless of the quality of the subsequent validation process.
Headspace GC-MS/MS: The Preferred Approach for Volatile Nitrosamines
For low-molecular-weight volatile nitrosamines such as NDMA, NDEA, NMEA, NDBA, NPYR, NPIP, and NDPA, headspace GC-MS/MS employing either chemical ionization (CI) or electron ionization (EI) with tandem mass spectrometric detection remains the regulatory-preferred analytical technique.
The headspace configuration offers several important advantages:
- It removes non-volatile matrix interferences originating from APIs, excipients, and residual solvents by analyzing only the vapor phase above the heated sample.
- It supports simultaneous determination of multiple nitrosamines, with published methods demonstrating the quantification of six to nine analytes within a single run.
- It enables sub-ppb sensitivity, with calibration ranges commonly spanning 2.5–40 ng/mL and limits of detection as low as 0.15–1.00 ng/mL for individual nitrosamines.
- It utilizes isotopically labeled internal standards such as d₆-NDMA and d₁₀-NDEA, which are added during sample preparation to monitor extraction efficiency and compensate for instrumental variability.
Critical headspace parameters, including equilibration temperature, equilibration time, vial pressure, and agitation frequency, must be optimized during method development and thoroughly documented. These variables are commonly evaluated as part of robustness studies during validation.
Compare Sample Introduction Techniques: Choosing the right extraction mode is critical for volatile compounds. Read our comparative guide on Direct Injection vs. Headspace Techniques for Nitrosamines.
LC-MS/MS and HPLC-HRMS for NDSRIs and Non-Volatile Nitrosamines
Nitrosamine Drug Substance-Related Impurities (NDSRIs) are structurally complex nitrosamines derived directly from the active pharmaceutical ingredient. Because these compounds are semi-volatile or non-volatile, they are generally unsuitable for headspace GC-MS/MS analysis.
Instead, reversed-phase or HILIC LC-MS/MS methods employing electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) are required.
Several important considerations influence LC-MS/MS method development:
- Chromatographic selectivity: The analytical method must effectively separate the NDSRI from the parent drug substance, metabolites, and degradation products. This can be particularly challenging when structural similarities are significant.
- Ionization mode selection: APCI frequently provides superior performance compared with ESI for certain NDSRI classes because it is less susceptible to matrix-induced ion suppression.
- Selected Reaction Monitoring (SRM): Two MRM transitions, consisting of a quantifier and a qualifier ion transition, are generally required to support unequivocal identification in GMP-regulated environments.
- High-resolution mass spectrometry: HPLC-QTOF and Orbitrap systems can support both targeted quantitation and untargeted screening for unknown nitrosamines within a single analytical workflow. These capabilities are especially valuable during risk assessments and confirmatory investigations.
Advanced MS Workflows: Learn how high-resolution instruments solve complex matrix challenges in our technical overview of HRMS for Nitrosamine Testing.
Regulatory Framework Governing Nitrosamine Method Validation
Nitrosamine method validation must satisfy the requirements of both ICH Q2(R2) and the nitrosamine-specific expectations established by ICH M7(R2), the FDA, EMA, and Health Canada. These requirements have undergone significant updates between 2023 and 2025.
Current Regulatory Framework
| Regulatory Body | Document | Key Analytical Requirement |
| ICH | M7(R2) | Defines nitrosamines as a cohort of concern and establishes AI-based limits |
| ICH | Q2(R2) | Defines analytical validation requirements and acceptance criteria |
| FDA | Control of Nitrosamine Impurities in Human Drugs, Revision 2 (September 2024) | LOQ must be at or below AI limits; validated method examples provided |
| EMA | NIOG Q&A Document (Revision 22+) | LOQ must be at or below reporting thresholds; confirmatory testing requirements aligned with ICH Q3A/B |
| USP | <1469> Nitrosamines in Drug Products | Provides a compendial framework for GC-MS/MS and LC-MS/MS methods |
| Health Canada | Nitrosamine Impurities in Pharmaceutical Drugs | Aligns with ICH M7(R2) and outlines submission expectations for Canadian regulatory filings |
One important regulatory distinction should be noted. EMA guidance specifies that when nitrosamines are controlled under ICH Q3A/B principles, the analytical method LOQ should be at or below the applicable reporting threshold. However, when nitrosamines are managed under the ICH M7 “cohort of concern” framework, the LOQ must be aligned with acceptable intake limits, resulting in a significantly more stringent requirement.
Stay Current with Evolving Frameworks: Global standards are shifting rapidly. Review the latest compliance impacts in our article on the Impact of ICH M7(R2) Updates on Nitrosamine Risk Assessment.
What Full GMP Validation of a Nitrosamine Method Actually Requires
Comprehensive GMP validation of a nitrosamine analytical method under ICH Q2(R2) requires evaluation of eight core validation parameters. Each parameter carries nitrosamine-specific expectations that are often more demanding than those applied to conventional impurity methods.
Complete Validation Parameter Requirements for Nitrosamine Methods
| Validation Parameter | Acceptance Criteria (Nitrosamine-Specific) | Importance |
| Specificity/Selectivity | No interference from blanks, placebo matrices, related substances, or reagents at the LOQ level | Ensures observed nitrosamine signals are genuine and not false positives |
| Linearity | R² ≥ 0.990 from LOQ to 120–150% of specification limit; y-intercept ≤2% of response at 100% specification | Demonstrates proportional response throughout the analytical range |
| LOD | ≤ one-third of the LOQ; signal-to-noise ratio ≥3:1 | Supports investigations below specification limits |
| LOQ | ≤ AI limit (target ≤30% AI); S/N ≥10:1; accuracy 70–130%; precision ≤25–30% RSD | Establishes the lowest reliably quantifiable concentration |
| Accuracy (Recovery) | 70–130% at LOQ; 80–120% at low, medium, and high levels; minimum of five replicates per level | Must be demonstrated within the actual product matrix |
| Repeatability | ≤25% RSD at LOQ; ≤15% RSD at higher concentrations | Evaluates intra-day performance under consistent conditions |
| Intermediate Precision | ≤25–30% RSD at LOQ; ≤20% RSD at higher levels; assessed across different analysts, days, or instruments | Identifies potential method variability |
| Robustness | Response remains within ±5% when key parameters are intentionally varied | Supports routine GMP implementation |
| Matrix Effect (LC-MS/MS) | Internal-standard-corrected matrix factor between 0.85 and 1.15 | Essential for ESI-based methodologies |
Matrix-effect assessment requires special attention. LC-MS/MS methods developed for NDSRIs are particularly vulnerable to ion suppression caused by co-eluting compounds originating from excipients, APIs, or residual solvents. Published studies involving rifampin and rifapentine NDSRIs have shown that without appropriately selected deuterated internal standards, ion suppression can reduce recoveries well below the acceptable 70–130% range despite apparently acceptable chromatographic performance.
Need Help Defending Specifications? Establishing validation criteria requires deep regulatory context. Learn how we assist with Nitrosamine Specification Setting to meet stringent reviewer demands.
The Staged Validation Pathway — From Initial Development to GMP Readiness
Most laboratories should not begin with full GMP validation. Instead, a phased development strategy allows analytical methods to mature efficiently while meeting regulatory expectations at each stage.
Stage 1: Risk Assessment-Guided Screening Method
The nitrosamine risk assessment defines the initial analyte list before laboratory work begins. Screening methods are designed primarily for broad detection and rapid evaluation rather than regulatory compliance.
Key characteristics include:
- Broad-spectrum multi-nitrosamine screening panels containing approximately 9–13 analytes.
- No formal validation beyond basic system suitability requirements.
- Qualitative or semi-quantitative results used to prioritize analytes for further investigation.
Stage 2: Fit-for-Purpose Confirmatory Method
After target nitrosamines have been identified, a confirmatory method is developed with:
- Demonstrated specificity.
- LOQ at or below the applicable AI limit.
- Accuracy and precision assessed at the LOQ and two additional concentration levels.
- Intermediate precision evaluated using multiple analysts or multiple analytical days.
The resulting data are generally sufficient to support regulatory notifications and interim control strategies.
Stage 3: Full GMP Method Validation
Full validation becomes necessary before analytical results can support:
- Batch release testing.
- Pharmacopoeial compliance testing.
- Stability studies.
- Regulatory submissions.
This stage includes:
- Complete characterization of all eight ICH Q2(R2) validation parameters.
- Preparation of a formal validation report with statistical analyses.
- Transfer of the validated method into the quality system through SOPs and analytical specifications.
- Establishment of system suitability criteria for ongoing performance monitoring.
Developing Custom Methods from Scratch? A phased, structured workflow is the most reliable way to avoid validation failures. Read our step-by-step guide to GC-MS Method Development for Nitrosamine Testing.
Internal Standard Selection — A Critical Determinant of Data Quality
The internal standard should be structurally identical or nearly identical to the target analyte, co-elute chromatographically with the analyte, and demonstrate effective correction of matrix-related response variation within the specific pharmaceutical matrix under investigation.
For small-molecule nitrosamines analyzed by GC-MS/MS or LC-MS/MS, isotopically labeled analogs represent the accepted industry standard:
- d₆-NDMA for NDMA quantification.
- d₁₀-NDEA for NDEA quantification.
- d₁₄-NDBA for NDBA quantification.
- d₈-NPYR for NPYR quantification.
For NDSRIs, ^13C- or ^15N-labeled structural analogs are generally preferred over partially deuterated compounds when isotopic exchange sites are present. Published studies involving cyclophosphamide-related nitrosamine impurities have demonstrated that deuterated internal standards that fail to co-elute precisely with the analyte, even by a few seconds, may inadequately compensate for ion suppression and produce systematic analytical bias that becomes apparent only during full validation.
Common Method Development Failures and Strategies to Prevent Them
Most nitrosamine validation failures originate from decisions made during method development rather than during the validation process itself.
LOQ Established Too Close to the AI Limit
Setting the LOQ directly at the AI limit leaves little room for analytical variability during routine testing. A target LOQ of ≤30% of the AI limit provides a more robust analytical margin.
Failure to Evaluate Matrix Effects
Clean chromatographic profiles do not guarantee the absence of ion suppression. Matrix factor studies should always be performed with and without internal standard correction.
Inappropriate Internal Standard Selection for NDSRIs
Using a generic nitrosamine internal standard, such as d₆-NDMA, for an NDSRI that elutes in a completely different chromatographic region provides minimal correction for matrix effects and can compromise data integrity.
Single-Level Accuracy Assessment
Regulatory reviewers frequently identify deficiencies when accuracy studies are performed at only one concentration level. Accuracy should be demonstrated across at least three concentration levels spanning the intended analytical range, including the LOQ.
Lack of Intermediate Precision Evaluation
Repeatability data collected on a single day are insufficient for full GMP validation. Intermediate precision studies should include multiple analysts and/or non-consecutive analytical days.
Inadequate Robustness Characterization
For headspace GC-MS/MS methods, equilibration temperature and equilibration time represent critical method variables. Analytical methods that have not been challenged with temperature variations of approximately ±5°C around the target equilibration temperature often encounter problems during routine GMP operation.
Managing Complex NDSRIs? When dealing with complex amine-derived impurities where limits are not pre-defined, check out our guide on the Nitrosamine CPCA Approach for NDSRIs.
Conclusion: From LOQ Design to GMP Submission — Achieving Analytical Excellence at Every Stage
Successful Nitrosamine Method Development and Validation Services rely on a combination of regulatory expertise, advanced analytical knowledge, and rigorous scientific execution. Establishing a scientifically justified LOQ relative to acceptable intake limits, selecting the most appropriate analytical platform for each nitrosamine class, evaluating matrix effects using suitable isotopically labeled internal standards, and fully addressing every ICH Q2(R2) validation requirement are not simply regulatory obligations. They are fundamental scientific requirements that determine whether a submission proceeds smoothly or attracts regulatory scrutiny.
At ResolveMass Laboratories Inc., our analytical scientists possess extensive hands-on experience at the intersection of ICH M7(R2) and ICH Q2(R2), supporting projects ranging from first-principles method development for novel NDSRIs to complete GMP validation programs prepared for CTD submission. We operate within a GMP-quality framework supported by certified reference standards, validated analytical methods, documented SOPs, and comprehensive regulatory-ready validation reports.
Frequently Asked Questions on Nitrosamine Method Development and Validation
The FDA does not prescribe a universal LOQ value for all nitrosamine methods. Instead, the LOQ must be sufficiently sensitive to ensure reliable measurement of nitrosamines at their applicable acceptable intake (AI) limits. In practice, most laboratories establish an LOQ at or below 30% of the specification limit, while highly sensitive methods often target 10% or less of the AI threshold. This approach provides additional confidence during routine quality control and regulatory inspections.
A single GC-MS/MS method is generally not suitable for analyzing both volatile nitrosamines and Nitrosamine Drug Substance-Related Impurities (NDSRIs). Volatile nitrosamines such as NDMA and NDEA are well suited for headspace GC-MS/MS analysis due to their physicochemical properties. In contrast, NDSRIs are typically larger, less volatile compounds that require LC-MS/MS techniques using reversed-phase or HILIC chromatography. Attempting to analyze NDSRIs using GC-MS/MS can lead to poor analyte recovery and unreliable results.
USP <1469> serves as a valuable framework for nitrosamine testing and provides general guidance on analytical methodologies. However, regulatory agencies expect method validation to be performed within the actual drug product matrix under investigation. Parameters such as specificity, accuracy, precision, matrix effects, and LOQ performance must be demonstrated for the specific product being tested. Therefore, USP <1469> should be viewed as a reference point rather than a complete submission-ready validation package.
NDSRIs are nitrosamine impurities that originate directly from the active pharmaceutical ingredient through reactions involving nitrosating agents and susceptible amine-containing structures. Unlike classical nitrosamines such as NDMA or NDEA, NDSRIs are unique to individual drug substances and often possess more complex chemical structures. They do not have universally assigned acceptable intake limits. Instead, their toxicological risk must be assessed individually using approaches such as ICH M7 principles, CPCA methodologies, or available genotoxicity data.
Nitrosamine analysis is typically performed at extremely low concentration levels, often in the sub-ppm or trace range. At these levels, even minor variations in sample preparation, instrument performance, analyst technique, or environmental conditions can significantly influence results. Intermediate precision studies are designed to capture these sources of variability by evaluating performance across different analysts, instruments, or testing days. As a result, they often reveal weaknesses that are not evident during repeatability assessments conducted under identical conditions.
Matrix effects are evaluated by comparing the response of an analyte in a clean solvent with the response obtained when the same analyte is introduced into an extracted sample matrix. This comparison helps determine whether components within the matrix suppress or enhance ionization during mass spectrometric analysis. Analysts calculate both the matrix factor (MF) and internal-standard-normalized matrix factor (IS-MF) to quantify these effects. Acceptable results demonstrate that the internal standard effectively compensates for matrix-related signal variations.
High-resolution mass spectrometry (HRMS), including UHPLC-QTOF and Orbitrap-based systems, is widely regarded as the preferred technology for unknown nitrosamine screening. These platforms provide accurate mass measurements and advanced acquisition modes that enable the detection of unexpected nitrosamine species. HRMS can identify potential impurities based on characteristic fragmentation patterns and molecular mass information, making it particularly valuable during risk assessments and impurity investigations. It also supports both targeted and untargeted screening workflows within a single analytical platform.
According to ICH Q2(R2), linearity should be demonstrated using a minimum of five concentration levels covering the intended analytical range. For nitrosamine methods, this range typically extends from the LOQ to approximately 120–150% of the specification limit. The calibration model should demonstrate a strong correlation between concentration and response, generally with an R² value of 0.990 or higher. Consistent residual distribution across the range is also important to confirm reliable quantification.
Reference:
- U.S. Food and Drug Administration. Control of Nitrosamine Impurities in Human Drugs: Guidance for Industry (Revision 2). CDER, September 2024. https://www.fda.gov/media/141720/download
- International Council for Harmonisation (ICH). Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk: M7(R2). 2023. https://www.ich.org/page/m7-genetic-toxicology
- International Council for Harmonisation (ICH). Validation of Analytical Procedures: Q2(R2). 2023. https://www.ich.org/page/q2r2-validation-analytical-procedures
- European Medicines Agency (EMA). Questions and Answers for Marketing Authorisation Holders on the CHMP Opinion for the Article 5(3) Referral on Nitrosamine Impurities in Human Medicinal Products (Revision 22). EMEA/H/A5(3)/1490. https://www.ema.europa.eu
- United States Pharmacopeia (USP). <1469> Nitrosamines in Drug Products. USP-NF. https://www.usp.org
- Veeprho. EMA Update on Nitrosamine Guidelines 2024. August 2025. https://veeprho.com/ema-update-on-nitrosamine-guidelines/
- Szczepańska K., et al. Development of a Sensitive Screening Method for Simultaneous Determination of Nine Genotoxic Nitrosamines in Active Pharmaceutical Ingredients by GC-MS. Pharmaceutics, 14(10), 2191. 2022. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9603764/

