
Introduction
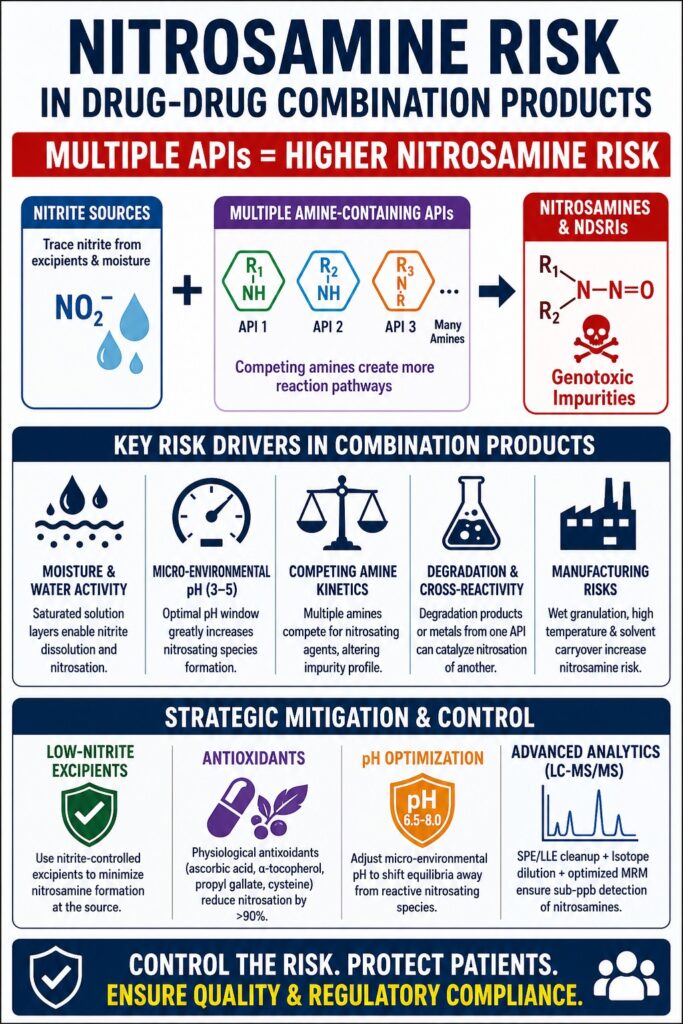
Nitrosamine Risk in Drug-Drug Combination Products has emerged as a significant toxicological challenge because combining multiple active pharmaceutical ingredients within a single formulation creates interconnected chemical pathways that can promote nitrosamine formation. When secondary or tertiary amines coexist with trace levels of nitrites in a shared micro-environment, their collective chemical interactions can substantially increase the likelihood of producing mutagenic impurities. Assessing and controlling this risk is considerably more demanding than evaluating single-active pharmaceutical ingredient (API) formulations. Each API contributes its own chemical reactivity, physicochemical characteristics, and degradation behavior, and their coexistence within a solid dosage matrix can influence essential factors such as micro-environmental pH, moisture absorption, and reaction kinetics. As a result, regulatory authorities worldwide now expect far more comprehensive chemical risk assessments, requiring manufacturers of fixed-dose combinations (FDCs) to implement formulation-specific evaluation and testing strategies.
To understand the fundamental chemistry behind these impurities, read our comprehensive guide on what are nitrosamines.
Meeting the rigorous Experience, Expertise, Authoritativeness, and Trustworthiness (E-E-A-T) expectations established by Google, while also satisfying international pharmaceutical regulatory standards, requires organizations to go beyond conventional compliance approaches. Advanced contract research organizations, including ResolveMass Laboratories Inc., utilize cutting-edge high-resolution tandem mass spectrometry (LC-MS/MS) to characterize complex multi-API formulations at sub-parts-per-billion (ppb) detection levels. By integrating comprehensive physicochemical characterization with advanced thermodynamic modeling, these laboratories identify potential cross-reactive pathways, helping manufacturers maintain formulation stability while safeguarding patient health.
Explore our specialized capabilities in nitrosamine testing in combination products to ensure full compliance for multi-API formulations.
Share via:
Article Summary:
- Multiple APIs significantly increase nitrosamine risk because they introduce interconnected chemical reactions where several amine-containing drug substances can compete for the same nitrosating agents, creating more complex impurity formation pathways than single-API formulations.
- Moisture, micro-environmental pH, and excipient-derived nitrites are the primary drivers of nitrosamine generation. Localized water layers inside tablets enable nitrite mobility, while acidic conditions and hygroscopic excipients accelerate the conversion of trace nitrites into reactive nitrosating species.
- Drug-drug interactions within fixed-dose combinations can alter impurity profiles through competing amine kinetics, cross-degradation, and catalytic reactions. As a result, the expected nitrosamine formed in one formulation may differ substantially from that in another, even when the same API is present.
- Manufacturing processes and formulation design strongly influence nitrosamine formation. Wet granulation, elevated processing temperatures, solvent carryover, packaging interactions, and moisture uptake during storage can all increase the likelihood of genotoxic impurity development.
- Comprehensive risk assessment requires orthogonal stress testing and advanced analytical characterization. Forced degradation studies combined with highly sensitive LC-MS/MS methods help identify potential Nitrosamine Drug Substance-Related Impurities (NDSRIs) and distinguish genuine impurities from complex formulation matrix interference.
- Targeted mitigation strategies can dramatically reduce nitrosamine formation. Using low-nitrite excipients, optimizing formulation pH, incorporating approved antioxidants, improving manufacturing controls, and applying isotope dilution LC-MS/MS provide effective approaches for minimizing impurity levels while maintaining product quality.
- Regulatory agencies now expect formulation-specific nitrosamine evaluations for combination products. Pharmaceutical manufacturers must adopt science-based risk assessments, robust analytical validation, and proactive formulation optimization to ensure long-term product stability, regulatory compliance, and patient safety.
Theoretical Kinetics and Chemical Drivers of Nitrosamine Risk in Drug-Drug Combination Products
The mechanisms responsible for amine nitrosation in combination drug products are controlled by competing chemical equilibria involving electrophilic species generated from nitrous acid and the unprotonated nucleophilic nitrogen atoms present in co-formulated active ingredients. The basicity of the amine (pKₐ), the surrounding micro-environmental pH, and the presence of competing amine-containing compounds collectively influence both the thermodynamic favorability and the kinetic rate of nitrosamine formation within the formulation matrix.
Aqueous nitrosation of secondary amines (R2NH) begins with the conversion of inorganic nitrite (NO2−), which is frequently introduced as a trace impurity from pharmaceutical excipients, into highly reactive electrophilic species. Under acidic micro-environmental conditions, typically within a pH range of 3 to 5, inorganic nitrite undergoes protonation to generate nitrous acid (HNO2):
NO2− + H+ ⇌ HNO2 (pKa ≈ 3.15)
Nitrous acid subsequently establishes equilibrium with several potent nitrosating species, most notably dinitrogen trioxide (N2O3), nitrosyl halides (NOX, such as ClNO), and the nitrous acidium ion (H2NO2+):
2HNO2 ⇌ N2O3 + H2O
HNO2 + H+ + Cl− ⇌ ClNO + H2O
These electrophilic intermediates preferentially react with the unprotonated, or free-base, form of the amine rather than its protonated ammonium counterpart ((\text{R}_2\text{NH}_2^+)), which is largely unreactive toward nitrosation. The concentration of the reactive unprotonated amine depends on both the local micro-environmental pH and the intrinsic basicity (pKₐ) of the specific amine group:
[R2NH] = [Amine]total 1 + 10(pKa − pH)
Since the free-base amine functions as the active nucleophile, highly basic aliphatic secondary amines (pKₐ ≈ 10–11) remain almost completely protonated under acidic conditions, leaving only a very small fraction available in the reactive form. Nevertheless, the free-base molecules that are present react extremely rapidly with N2O3, exhibiting rate constants that approach the diffusion-controlled limit of approximately 109 M−1s−1. In contrast, weakly basic amines, including heterocyclic secondary amines and aryl-alkyl secondary amines with pKₐ values below 5, exist predominantly in their unprotonated state under the same acidic conditions. Consequently, the nitrosation susceptibility of a particular API cannot be predicted solely from its chemical structure. Instead, it depends on the continuously changing balance between pH and the concentration of the reactive free-base amine present within the formulation.
Discover the underlying synthesis risks and reaction mechanisms in our analysis of nitrosamine formation pathways API synthesis.
Saturated Solution Layers and Water Activity in Solid Dosage Forms
Nitrosamine formation in solid oral dosage forms does not occur primarily through direct solid-state reactions. Instead, it takes place within localized saturated solution layers that develop from bound or absorbed moisture inside the tablet matrix. Within these microscopic liquid domains, localized water activity ((a_w)) and thermodynamic equilibrium govern the conversion of nitrite impurities into highly reactive nitrosating species such as dinitrogen trioxide (N2O3).
For nitrosation to proceed in a solid pharmaceutical product, moisture serves as the essential medium that enables both the dissolution and mobility of inorganic nitrites and amine-containing precursors. Saturated solution layers typically develop at the interfaces between hygroscopic excipients and API crystals. Inside these confined liquid regions, the relative humidity within the package or moisture absorbed from the surrounding environment—whether during wet granulation or prolonged storage under elevated humidity—determines the amount of water available to dissolve and transport the reactive components.
Experimental studies have demonstrated that even in formulations lacking nitrosatable amines, inorganic nitrites remain chemically unstable under humid storage conditions ranging from (25°C/60% RH) to (40°C/75% RH). Under these conditions, nitrite concentrations may decline by as much as 95% within 30 days due to the volatilization of nitrous acid-derived gases, including N2O3 and N2O4, as well as O-nitrosation reactions involving hydroxyl-containing excipients such as microcrystalline cellulose and mannitol. However, when a nitrosatable API is present within the same humid and mildly acidic matrix, the reactive nitrosating species are rapidly consumed by dissolved amines, resulting in the formation and retention of nitrosamine impurities within the drug product.
Competing Amine Paradigm and Nitrosamine Risk in Drug-Drug Combination Products
In pharmaceutical formulations containing multiple active pharmaceutical ingredients, competing amine kinetics determine which API preferentially reacts with the available nitrosating species according to its nucleophilicity, concentration, and protonation state. Highly basic secondary aliphatic amines continuously compete with less basic heterocyclic amines, causing the anticipated nitrosamine drug-substance related impurity (NDSRI) profile to evolve throughout the product’s shelf life.
When several amine-containing APIs or trace processing impurities coexist within a single formulation, available nitrosating agents are simultaneously consumed through multiple competing reaction pathways. The relative rates of nitrosamine formation for individual amines can be described using concentration-dependent kinetic relationships:
d[Nitrosamine A]/dt d[Nitrosamine B]/dt = kA [Amine A]unprotonated kB [Amine B]unprotonated
In a fixed-dose combination containing a highly basic drug substance, such as a beta-blocker with a pKₐ of approximately 9.5, together with a less basic but highly soluble compound such as metformin, the local micro-environmental pH ultimately determines which molecule undergoes nitrosation preferentially. When formulation adjustments lower the local pH to improve the stability of the beta-blocker, metformin may instead function as a kinetic sink because of its substantially higher molar concentration. This shift redirects nitrosation toward smaller nitrosated byproducts rather than producing the NDSRI that would otherwise be expected.
Learn how complex drug-derived impurities behave differently from simpler compounds in our evaluation of ndsri vs simple nitrosamines.
Orthogonal Forced Degradation and Structural Susceptibility of Complex Amines
Determining the structural susceptibility of complex active pharmaceutical ingredients (APIs) to nitrosation requires orthogonal forced degradation studies specifically designed to define the chemical and physical boundaries of nitrosamine formation. These standardized stress-testing protocols evaluate whether structurally complex secondary or tertiary amines are capable of forming stable nitrosamine drug-substance related impurities (NDSRIs) under worst-case manufacturing, storage, and environmental conditions.
To comprehensively assess the possibility of generating novel N-nitrosamines from structurally diverse drug substances, current regulatory frameworks recommend a standardized set of three complementary orthogonal forced nitrosation conditions. These analytical stress studies reproduce severe chemical environments that may arise during API synthesis, drug product formulation, or even gastrointestinal exposure. The resulting data provide a scientifically sound basis for nitrosamine risk assessment and impurity classification.
Aqueous Acidic Nitrosation:
In this approach, the amine is treated with an excess of sodium nitrite in a strongly acidic aqueous solution (pH ≤ 2) at ambient temperature. These conditions maximize the formation of highly reactive nitrosating species, particularly the nitrous acidium ion (H2NO2+) and nitrous acid, thereby establishing the maximum theoretical susceptibility of the amine toward nitrosation.
Organic/Non-Aqueous Nitrosation:
This experiment is performed in an organic solvent, such as dichloromethane or acetonitrile, using organic nitrosating reagents including t-butyl nitrite or amyl nitrite. The non-aqueous environment enables the evaluation of highly lipophilic amines that may exhibit limited solubility or reduced reactivity in aqueous systems, while also representing potential risks associated with residual synthetic solvents carried over from manufacturing.
Phase-Transfer/Catalyzed Nitrosation:
Nitrosation is examined within a biphasic or heterogeneous reaction system containing formaldehydes, transition metals, or phase-transfer catalysts under mildly acidic to neutral conditions. This orthogonal stress model more closely represents complex pharmaceutical formulations in which excipients, packaging-derived leachables, or catalytic impurities may accelerate nitrosamine formation through chemical or physical interactions.
The successful synthesis and analytical isolation of an NDSRI reference standard under these forced degradation conditions are essential for reliable analytical characterization. Conversely, if comprehensive orthogonal studies consistently demonstrate that a particular complex amine cannot be converted into its corresponding N-nitrosamine because of steric hindrance, competing intramolecular reaction pathways, or the thermodynamic instability of the nitroso derivative, these findings provide robust scientific evidence to support a reduced routine testing strategy in accordance with EMA and FDA regulatory expectations.
Read about the toxicological frameworks governing these assessments in our article on genotoxic impurity testing ich m7 nitrosamines.

Unique Risk Drivers Amplifying Nitrosamine Risk in Drug-Drug Combination Products
The factors responsible for increasing Nitrosamine Risk in Drug-Drug Combination Products primarily arise from excipient-derived nitrite cross-reactivity, catalyzed degradation pathways between co-formulated APIs, and shared moisture adsorption within the formulation. Collectively, these mechanisms generate highly reactive micro-environments during wet granulation, tablet compression, and long-term storage, significantly increasing the probability of nitrosamine formation.
To effectively control these risks, researchers must identify and evaluate the formulation variables that distinguish single-API products from fixed-dose combination formulations.
| Risk Factor | Impact in Single-API Formulations | Impact in Multiple-API Combination Products |
|---|---|---|
| Excipient Nitrites | Trace nitrites typically react with a single amine-containing API through a relatively straightforward reaction pathway. | Trace nitrites can simultaneously interact with multiple competing amine-containing APIs, creating bidirectional or multidirectional reaction pathways that substantially increase nitrosamine formation potential. |
| Degradation Kinetics | Degradation generally follows predictable pathways governed by the thermodynamic properties of a single API. | Cross-degradation mechanisms become significant, where degradation products or transition metals generated from one API may catalyze nitrosation reactions involving the companion API. |
| Micro-environmental pH | Formulation pH is optimized to preserve API stability while minimizing nitrosation risk. | Adjustments made to stabilize one API may unintentionally shift the neighboring API into its optimal nitrosation window (pH 3–5), thereby increasing impurity formation. |
| Moisture & Granulation | Moisture introduced during wet granulation briefly supports localized solution-state reactions around a single API. | Hygroscopic behavior of one API may continuously attract environmental moisture, sustaining long-term solution-state nitrosation reactions involving adjacent APIs throughout storage. |
| Packaging & Migration | Packaging-derived nitrosamines or nitrocellulose-related impurities generally interact with only one active ingredient. | Volatile secondary amines and nitrite-containing packaging leachables may simultaneously react with multiple APIs, substantially increasing the overall nitrosamine impurity burden. |
Granulation and Physical Manufacturing Risks
The manufacturing processes used for multi-API oral solid dosage forms represent critical stages during which nitrosamine formation can be initiated. Among these operations, wet granulation is considered one of the highest-risk processing steps. The intentional addition of water, combined with elevated temperatures encountered during fluid-bed drying, creates an optimal kinetic environment for solution-state nitrosation before the formulation has completely dried.
When one of the co-formulated APIs possesses high aqueous solubility, it readily dissolves during wet granulation, producing a concentrated liquid phase capable of rapidly dissolving and interacting with trace nitrites originating from excipients. By contrast, manufacturing approaches such as dry granulation (roller compaction) or direct compression eliminate this transient liquid phase, thereby significantly reducing opportunities for nitrite-to-nitrosamine conversion.
In addition, combination product manufacturing presents an increased risk of cross-contamination within shared production facilities. The reuse of recovered solvents—including DMF, THF, or toluene—that have not been subjected to rigorous purification procedures may introduce residual secondary amines or nitrosating species from previous manufacturing campaigns. Such carryover contaminants can subsequently react within the new formulation, leading to the formation of genotoxic nitrosamine impurities in the finished combination product.
Case-Based Evidence: High-Scrutiny Fixed-Dose Combination Products
Evidence from regulatory recalls, post-market investigations, and long-term stability studies has demonstrated that several widely prescribed fixed-dose combination products—including formulations containing metformin, sitagliptin, vildagliptin, bisoprolol, or hydrochlorothiazide—are particularly susceptible to nitrosamine formation. These real-world examples highlight how elevated API concentrations, shared excipient systems, and complex formulation micro-environments can collectively promote the simultaneous formation of both low-molecular-weight nitrosamines and structurally complex nitrosamine drug-substance related impurities (NDSRIs).
Metformin and DPP-4 Inhibitor Co-formulations
Fixed-dose antidiabetic formulations that combine high-dose metformin with DPP-4 inhibitors such as sitagliptin or vildagliptin are particularly susceptible to dual nitrosation pathways. These formulations can generate both low-molecular-weight nitrosamines, including NDMA, and complex nitrosamine drug-substance related impurities (NDSRIs), such as NTTP and N-nitroso-vildagliptin. Owing to its highly hydrophilic nature and high formulation dose, metformin acts as an efficient moisture-retaining matrix, substantially increasing the rate at which sitagliptin-related impurities are converted into NTTP.
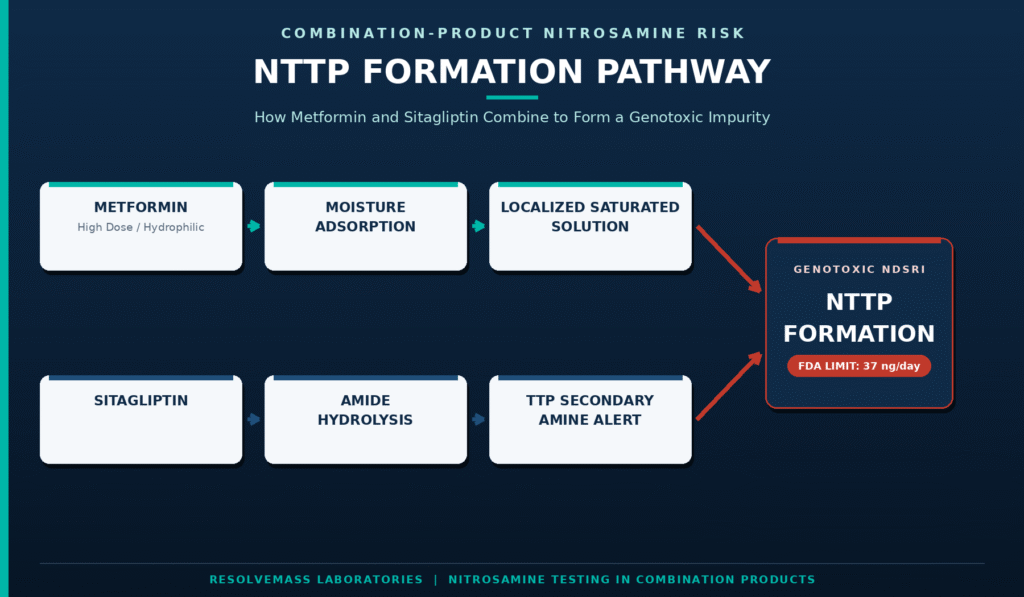
Metformin (High Dose/Hydrophilic) ──> Moisture Adsorption ──> Localized Saturated Solution
│
Sitagliptin ──> Amide Hydrolysis ──> TTP Secondary Amine Alert ─────┴─> NTTP Formation
Within sitagliptin/metformin fixed-dose combination tablets, including generic equivalents of Janumet®, the secondary amine impurity triazolopyrazine (TTP) may originate either as a residual synthetic intermediate or develop gradually through amide hydrolysis of sitagliptin during storage. This impurity readily reacts with trace nitrites originating from pharmaceutical excipients, resulting in the formation of the highly potent NDSRI known as NTTP. Owing to its significant toxicological concern, the FDA has established an exceptionally stringent acceptable intake limit of only 37 ng/day for NTTP, corresponding to an analytical specification of approximately 0.37 ppm for a 100 mg sitagliptin dose.
Because metformin is commonly administered at daily doses reaching 2000 mg within the same tablet matrix, its physicochemical characteristics largely determine the local micro-environmental water activity ((a_w)) of the finished dosage form. The hygroscopic nature of metformin promotes moisture uptake throughout storage, facilitating the dissolution and transport of trace nitrite impurities present in commonly used excipients such as povidone (PVP) and croscarmellose sodium. These conditions significantly accelerate NTTP formation during the commercial shelf life of the product.
An analogous concern exists in vildagliptin/metformin combination tablets. The secondary pyrrolidine amino group present in vildagliptin undergoes direct N-nitrosation, producing the NDSRI N-nitroso-vildagliptin, which must be controlled within a strict specification limit of 15 ppm. During formulation development and stability studies, competitive nitrosation reactions occur between the secondary amine of vildagliptin and trace amine impurities associated with metformin, including dimethylamine. This interaction creates a dual-impurity scenario in which both NDMA and N-nitroso-vildagliptin may be generated simultaneously, necessitating dual-channel LC-MS/MS surveillance for comprehensive impurity monitoring.
For detailed testing considerations regarding these anti-diabetic medications, see nitrosamine testing for a metformin generic.
Bisoprolol and Hydrochlorothiazide Cardiovascular Combinations
Cardiovascular fixed-dose combination tablets containing bisoprolol and hydrochlorothiazide present a particularly challenging nitrosamine risk profile because they are capable of generating two separate genotoxic impurities: N-nitroso-bisoprolol and N-nitroso-hydrochlorothiazide. The inherent chemical instability of N-nitroso-hydrochlorothiazide enables its decomposition to release highly reactive nitrosyl cations, which subsequently promote cross-nitrosation reactions involving bisoprolol.
Hydrochlorothiazide (HCTZ) contains a cyclic secondary sulfonamide functional group that readily undergoes nitrosation under mildly acidic conditions, producing the NDSRI N-nitroso-hydrochlorothiazide (NNO-HCTZ). Physicochemical investigations have demonstrated that NNO-HCTZ is highly unstable in aqueous environments with pH values exceeding 5, where it rapidly decomposes into formaldehyde, thiatriazine, and aminobenzenesulfonic acid. Under acidic formulation or gastric conditions, typically between pH 1 and 5, decomposition occurs more gradually but continuously releases highly reactive nitrosyl cations (NO+).
When HCTZ is co-formulated with the beta-blocker bisoprolol, which contains a highly reactive secondary aliphatic amine, decomposition of NNO-HCTZ serves as an in situ source of active nitrosyl species. This internal “nitroso transfer” mechanism eliminates dependence on external nitrite impurities and promotes rapid catalytic cross-nitrosation of bisoprolol, leading to the formation of NNO-Bisoprolol. Such synergistic chemical interactions create a high-risk impurity pathway that cannot be accurately predicted by evaluating hydrochlorothiazide or bisoprolol as individual monotherapy products.
Review our technical breakdown of cardiovascular risk screening at nitrosamine testing beta blockers.
Analytical Methodologies for Resolving Matrix Interference in Combination Testing
Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) remains the analytical gold standard for detecting and quantifying trace nitrosamines in highly complex combination drug products. Achieving reliable detection at sub-parts-per-billion (ppb) concentrations requires highly selective chromatographic separation together with rigorously validated analytical methods capable of minimizing matrix-induced ion suppression.
Quantification of nitrosamines at picogram-per-milliliter (pg/mL) concentrations in the presence of co-eluting APIs present at milligram-per-milliliter (mg/mL) levels is significantly complicated by matrix effects. These effects occur when excipients, degradation products, or high concentrations of drug substances interfere with the ionization efficiency of the target nitrosamine within the ion source of the mass spectrometer. Matrix effects can be expressed using the following relationship:
Matrix Effect (%) = Peak Area of Analyte in Spiked Extracted Matrix Peak Area of Analyte in Pure Solvent × 100
Matrix effect values below 100% indicate ion suppression, whereas values above 100% indicate ion enhancement. Because these phenomena can significantly compromise analytical accuracy in multi-API formulations, advanced sample preparation techniques are essential.
Solid-Phase Extraction (SPE):
The use of mixed-mode or specialized copolymeric SPE cartridges enables selective retention of polar nitrosamines while effectively removing highly concentrated APIs and hydrophobic excipients that contribute to matrix interference.
Liquid-Liquid Extraction (LLE):
Carefully selected organic solvents, including dichloromethane, facilitate selective extraction of low-molecular-weight nitrosamines from complex aqueous tablet extracts while minimizing co-extraction of interfering formulation components.
Isotope Dilution-Mass Spectrometry (IDMS):
Samples are fortified with stable isotopically labeled internal standards, commonly deuterium ((^2\text{H}))- or carbon-13 ((^{13}\text{C}))-labeled analogs such as NDMA-d6 or N-nitroso-bisoprolol-d5. Because the labeled compounds experience the same extraction efficiency and ionization suppression as their native counterparts, they effectively compensate for analytical variability and significantly improve quantitative accuracy.
Read more about optimization workflows in our overview of nitrosamine method development and validation services.
Chromatographic and Mass Spectrometric Parameters for Double-NDSRI Quantitation
Simultaneous determination of multiple structurally distinct nitrosamines requires carefully optimized chromatographic gradients, highly selective stationary phases, and precisely optimized multiple reaction monitoring (MRM) transitions. Employing specialized stationary phases, including Biphenyl and C30 columns, minimizes co-elution with highly concentrated APIs while reducing isobaric solvent interferences.
To achieve complete chromatographic separation between high-concentration parent drug substances and trace polar nitrosamines such as NDMA—which frequently elutes near the solvent front on conventional reversed-phase columns—specialized stationary phases are essential. C30 columns provide enhanced shape selectivity together with stronger hydrophobic interactions under highly aqueous mobile-phase conditions, allowing complete resolution of polar nitrosamines from overloaded API peaks. Biphenyl stationary phases exploit strong (\pi-\pi) interactions, making them particularly suitable for separating structurally complex NDSRIs such as NNO-Bisoprolol and NNO-HCTZ.
Standardized LC-MS/MS methods for highly sensitive multi-analyte MRM quantification in combination products typically employ positive atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI). Representative MRM transition parameters are summarized below.
| Analyte | Parent Ion (m/z) | Product Ion (m/z) | Quantifier / Qualifier | Ionization Mode | Collision Energy (eV) |
|---|---|---|---|---|---|
| NDMA | 75.05 | 58.05 | Quantifier | APCI⁺ | 9 |
| NDMA | 75.05 | 43.00 | Qualifier | APCI⁺ | 13 |
| NNO-Bisoprolol | 377.10 | 275.15 | Quantifier | ESI⁺ | -22 |
| NNO-Bisoprolol | 377.10 | 246.10 | Qualifier | ESI⁺ | -18 |
| NNO-HCTZ | 325.00 | 293.95 | Quantifier | ESI⁻ | 24 |
| NTTP (N-Sitagliptin) | 437.15 | 235.10 | Quantifier | APCI⁺ | 15 |
Strategic Mitigation of Nitrosamine Risk in Drug-Drug Combination Products and Advanced Bioequivalence Approaches
Effective mitigation of Nitrosamine Risk in Drug-Drug Combination Products requires a proactive formulation strategy that incorporates scientifically designed product modifications, including the use of physiological antioxidants and micro-environmental pH regulators. When formulation changes are implemented to minimize nitrosamine generation, alternative bioequivalence (BE) approaches provide scientifically justified pathways for demonstrating therapeutic equivalence while preserving the absorption characteristics of the drug product.
If confirmatory analytical testing demonstrates that a multi-API formulation exceeds the established regulatory acceptable intake limits for nitrosamines or nitrosamine drug-substance related impurities (NDSRIs), manufacturers must introduce formulation-level interventions to suppress nitrosation reactions. Extensive FDA-supported research has shown that incorporating small physiological quantities of naturally occurring antioxidants, including ascorbic acid, α-tocopherol, propyl gallate, or cysteine, directly into the tablet formulation can reduce nitrosamine formation by more than 90%. These antioxidants function as competitive scavengers by rapidly converting reactive nitrosating species, such as nitrous acid, into relatively unreactive nitric oxide ((\text{NO})) before they can react with susceptible secondary or tertiary amine groups present within the co-formulated APIs.
To learn more about formulation-level stabilization, explore our nitrosamine reformulation strategy
and our comprehensive nitrosamine control strategy development services.
In addition, adjusting the micro-environmental slurry pH of the formulation through the controlled addition of organic acids or basic amino acids can significantly influence the protonation states of both nitrite species and competing amines. Under certain conditions, lowering the local pH promotes the rapid degradation and volatilization of inorganic nitrites through conversion into gaseous nitrogen oxides. Conversely, increasing the slurry pH to a mildly alkaline range (pH 6.5–8.0) shifts the equilibrium between nitrite and nitrous acid away from the reactive protonated electrophilic species, thereby interrupting the sequence of reactions responsible for nitrosamine formation.
Navigating Alternative Bioequivalence (BE) Frameworks During Reformulation
When antioxidants, pH modifiers, or other formulation changes are introduced to reduce nitrosamine formation, the resulting product is classified as a reformulated dosage form. Rather than undertaking expensive and time-consuming in vivo clinical bioequivalence studies, manufacturers may utilize alternative science-based BE strategies described in the latest FDA Guidance (September 2024, Revision 2) to demonstrate therapeutic equivalence.
Biopharmaceutics Classification System (BCS) Class I and Class III Exceptions:
If the APIs within the combination product are classified as BCS Class I (high solubility and high permeability) or BCS Class III (high solubility and low permeability), regulatory guidance permits the incorporation of limited quantities of approved antioxidants, including ascorbic acid or propyl gallate, without requiring in vivo bioequivalence studies. This approach is acceptable provided that the cumulative antioxidant exposure remains below 10 mg/day and all other regulatory requirements are satisfied.
In Vitro Transporter and Permeability Verification:
Comprehensive Caco-2 cell permeability investigations have demonstrated that trace concentrations of antioxidants such as ascorbic acid, cysteine, and α-tocopherol do not significantly alter membrane permeability or the activity of important intestinal transporters, including P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP), for representative BCS Class III compounds. Manufacturers may therefore submit validated in vitro dissolution profiles together with Caco-2 permeability data to demonstrate that the reformulated product maintains equivalent absorption characteristics for each active pharmaceutical ingredient.
SUPAC Scale-Up and Post-Approval Changes Alignment:
When formulation modifications—such as replacing conventional microcrystalline cellulose with a certified low-nitrite grade or eliminating a glidant to optimize powder flow—remain within Level 1 or Level 2 SUPAC-IR classifications, regulatory approval can generally be supported through comparative in vitro dissolution testing conducted in multiple dissolution media (pH 1.2, 4.5, and 6.8), eliminating the need for in vivo bioequivalence studies. More extensive Level 3 modifications, including changes to processing solvents or release-controlling excipients, typically require a single-dose fasting bioequivalence study using the highest dosage strength.
Conclusions on Managing Nitrosamine Risk in Drug-Drug Combination Products
Successfully minimizing Nitrosamine Risk in Drug-Drug Combination Products requires a multidisciplinary strategy that integrates thermodynamic modeling, orthogonal stress testing, and advanced tandem mass spectrometry. Collaborating with specialized contract research organizations such as ResolveMass Laboratories Inc. enables pharmaceutical manufacturers to effectively evaluate complex multi-API formulations while meeting stringent international regulatory expectations.
As regulatory oversight continues to become more rigorous worldwide, evaluating nitrosamine risk on the basis of individual APIs alone is no longer sufficient. Combination drug products require product-specific analytical validation because of the increased likelihood of competitive nitrosation, cross-degradation mechanisms, and significant matrix-induced ion suppression during mass spectrometric analysis. By implementing advanced chromatographic separation techniques, applying stable isotope dilution methodologies, and proactively redesigning formulations through the use of low-nitrite excipients and protective antioxidants, manufacturers can strengthen product quality and support long-term regulatory compliance. Partnering with experienced analytical organizations such as ResolveMass Laboratories Inc. provides the scientifically robust, E-E-A-T-aligned evidence necessary to protect patient safety, maintain product quality, and ensure uninterrupted commercial supply.
For customized nitrosamine risk assessments, orthogonal stress testing, and validated LC-MS/MS quantification of combination drug products, contact the technical specialists at ResolveMass Laboratories Inc.:
Frequently Asked Questions
Metformin-sitagliptin fixed-dose combinations are considered high risk because both the chemical properties of the APIs and the formulation environment contribute to nitrosamine formation. Metformin is used at relatively high doses and readily absorbs moisture, increasing water activity within the tablet. At the same time, degradation of sitagliptin can generate reactive secondary amine intermediates that may react with trace nitrites present in excipients, increasing the likelihood of forming impurities such as NTTP during storage.
The competing amine kinetic paradigm describes a situation in which multiple amine-containing compounds within the same formulation compete for available nitrosating species. The reaction outcome depends on factors such as amine concentration, pKa, local pH, and chemical reactivity. As these variables change throughout manufacturing or storage, the dominant nitrosation pathway may also change, resulting in different nitrosamine impurity profiles over the product’s shelf life.
Antioxidants such as ascorbic acid, cysteine, propyl gallate, and α-tocopherol help reduce nitrosamine formation by reacting with nitrosating species before they reach susceptible amine groups. By scavenging these reactive intermediates, antioxidants interrupt the chemical sequence responsible for nitrosation. When carefully incorporated into a formulation, they can significantly reduce impurity formation while maintaining product stability and therapeutic performance.
In certain situations, manufacturers may not need to conduct new in vivo bioequivalence studies after reformulation. Current FDA guidance allows alternative approaches for eligible BCS Class I and Class III products when formulation changes are minor and scientifically justified. Supporting evidence typically includes comparative dissolution testing, permeability studies such as Caco-2 assays, and other validated in vitro data demonstrating that drug absorption remains unchanged.
Co-elution occurs when high concentrations of APIs and trace nitrosamines enter the mass spectrometer simultaneously, creating significant matrix interference. This often leads to ion suppression or ion enhancement, reducing analytical sensitivity and potentially affecting quantification accuracy. To overcome these challenges, laboratories optimize sample preparation, chromatographic separation, and internal standard strategies to ensure reliable detection of trace nitrosamine impurities.
N-nitroso-hydrochlorothiazide is considered particularly important because it can decompose under certain formulation or physiological conditions, releasing highly reactive nitrosating species. These reactive intermediates may subsequently interact with neighboring APIs that contain susceptible amine groups, promoting additional nitrosation reactions within the same dosage form. This secondary reaction pathway increases the overall complexity of nitrosamine risk assessment for combination products.
Orthogonal forced degradation studies expose APIs to multiple controlled stress conditions, including acidic, organic, and catalyst-assisted environments, to evaluate their ability to form nitrosamines. When repeated testing consistently demonstrates that a specific compound does not generate a stable NDSRI, the resulting data provide strong scientific evidence supporting a reduced testing strategy. These findings can help justify regulatory waivers where appropriate under established guidance.
The Carcinogenic Potency Categorization Approach (CPCA) provides a science-based framework for estimating acceptable intake limits for newly identified NDSRIs. It evaluates structural features, functional groups, and other molecular characteristics associated with carcinogenic potential. In combination products, CPCA helps toxicologists rapidly prioritize newly detected impurities and supports consistent risk assessments during regulatory submissions.
C30 and Biphenyl chromatographic columns offer improved selectivity for separating trace nitrosamines from highly concentrated APIs compared with conventional C18 columns. C30 stationary phases provide enhanced retention of small polar nitrosamines under highly aqueous conditions, while Biphenyl columns utilize aromatic π-π interactions to improve separation of structurally complex NDSRIs. These enhanced chromatographic properties minimize co-elution, reduce matrix interference, and improve the accuracy and sensitivity of LC-MS/MS analysis.
Reference:
- Cioc, R. C., Joyce, C., Mayr, M., & Bream, R. N. (2023). Formation of N-nitrosamine drug substance related impurities in medicines: A regulatory perspective on risk factors and mitigation strategies. Organic Process Research & Development, 27(10), 1736–1750. https://doi.org/10.1021/acs.oprd.3c00153
- Naiffer_Host. (2026, January 21). Nitrosamine control in metformin/vildagliptin DP. USP Nitrosamines Exchange. https://nitrosamines.usp.org/t/nitrosamine-control-in-metformin-vildagliptin-dp/15058
- Jireš, J., Douša, M., Gibala, P., & Kubelka, T. (2022). N-Nitrosation in the absence of nitrosating agents in pharmaceuticals? Journal of Pharmaceutical and Biomedical Analysis, 218, 114872. https://doi.org/10.1016/j.jpba.2022.114872
- Naiffer_Host. (2024, July 11). AAPS PharmSci360 – Nitrosamines. USP Nitrosamines Exchange. https://nitrosamines.usp.org/t/aaps-pharmsci360-nitrosamines/10657
- Scientific Committee on Consumer Safety (SCCS). (2012, March 27). Opinion on nitrosamines and secondary amines in cosmetic products (SCCS/1458/11). European Commission. https://ec.europa.eu/health/scientific_committees/consumer_safety/docs/sccs_o_090.pdf
- U.S. Food and Drug Administration. (2024, September). Control of nitrosamine impurities in human drugs: Guidance for industry (Revision 2). U.S. Department of Health and Human Services. https://www.fda.gov/media/187314/download
- U.S. Food and Drug Administration. (2024, September). Control of nitrosamine impurities in human drugs: Guidance for industry (Revision 2). U.S. Department of Health and Human Services. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/control-nitrosamine-impurities-human-drugs

