
Introduction
The critical operational and compliance difference between a nitrosamine screening method and a confirmatory method is primarily defined by their analytical specificity, sensitivity, and quantitative accuracy. These characteristics determine how mutagenic impurities are identified, assessed, and controlled under a comprehensive Nitrosamine Screening vs Confirmatory Method framework. In the current global regulatory environment governed by organizations such as the United States Food and Drug Administration (FDA), the European Medicines Agency (EMA), and Health Canada, pharmaceutical manufacturers are required to evaluate and control trace-level N-nitrosamines present in active pharmaceutical ingredients (APIs) and finished drug products.
N-nitrosamines are categorized under the International Council for Harmonisation (ICH) M7(R2) guideline as “cohort of concern” compounds. These substances are recognized as highly potent mutagenic carcinogens that may pose significant physiological risks even at extremely low concentrations. To manage this potential risk, regulatory authorities require a systematic, step-wise control strategy that includes document-based risk assessment, analytical testing, and appropriate risk mitigation measures.
: Learn more about navigating compliance frameworks with our complete guide on Genotoxic Impurity Testing and ICH M7 Nitrosamines Guidelines.
Although both screening and confirmatory testing are performed during the analytical testing stage, they differ significantly in terms of scientific approach, extraction procedures, validation requirements, and analytical instrumentation used to achieve regulatory compliance.
Developing a scientifically robust risk-control strategy requires a comprehensive understanding of these two analytical approaches. For contract research and development organizations operating under stringent regulatory standards, such as ResolveMass Laboratories Inc. in Laval, Québec, understanding the distinction between these analytical methods is essential for delivering reliable testing solutions. With a Health Canada Drug Establishment Licence (DEL 3-002945-A), United States FDA Registration (FEI 3042696771), and ISO 9001:2015 certification, ResolveMass applies these specialized method categories to support drug substances from initial risk evaluation through validated analytical testing and commercial-grade batch release.
Discover how our specialized teams can protect your product quality through comprehensive Nitrosamine Control Strategy Development Services.
This article examines the key chromatographic, mass spectrometric, and regulatory differences that distinguish nitrosamine screening methods from confirmatory methods.
Share via:
Article Summary:
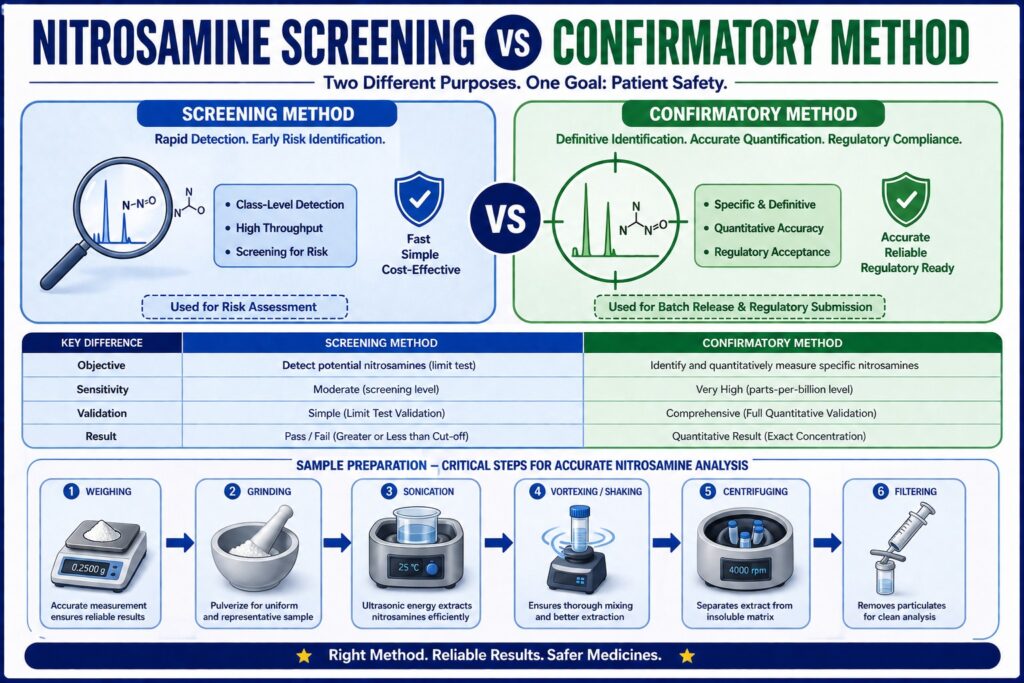
- Nitrosamine screening methods are designed for rapid, high-throughput detection of potential nitrosamine contamination. They provide an initial risk assessment by identifying samples that may require additional investigation rather than delivering precise quantitative results.
- Confirmatory methods are highly sensitive, fully validated analytical procedures used to accurately identify and measure individual nitrosamine impurities. These methods generate regulatory-compliant data suitable for product release and official submissions.
- The main distinction lies in their purpose: screening methods focus on early detection and efficient sample evaluation, while confirmatory methods provide definitive identification, exact quantification, and compliance with regulatory expectations.
- Advanced analytical technologies such as LC-MS/MS, GC-MS/MS, LC-HRMS, and isotope-labeled internal standards are commonly employed in confirmatory testing to achieve parts-per-billion sensitivity, excellent specificity, and reliable quantitative accuracy.
- Regulatory agencies, including the FDA, EMA, Health Canada, and ICH, require comprehensive validation of confirmatory methods to demonstrate accuracy, precision, specificity, robustness, and suitable limits of quantitation for trace-level nitrosamine analysis.
- Proper sample preparation plays a critical role in analytical performance. Steps such as weighing, grinding, sonication, vortex mixing, centrifugation, and filtration help improve extraction efficiency, minimize matrix interference, and ensure reproducible analytical results.
- A comprehensive nitrosamine control strategy combines rapid screening with confirmatory analysis, enabling pharmaceutical manufacturers to detect potential risks early, verify impurity levels accurately, support regulatory compliance, and maintain product quality throughout development and commercial manufacturing.
Core Analytical Principles of Nitrosamine Screening Methods
Nitrosamine screening methods are rapid and high-throughput analytical approaches developed to detect the potential presence of nitroso-compounds or specific target nitrosamine impurities at or above a predefined level of concern. These screening techniques function primarily as binary or semi-quantitative assessment tools, enabling quality control teams to efficiently differentiate between compliant batches and samples requiring further investigation due to potential mutagenic contamination.
Nitrosamine Screening Workflow
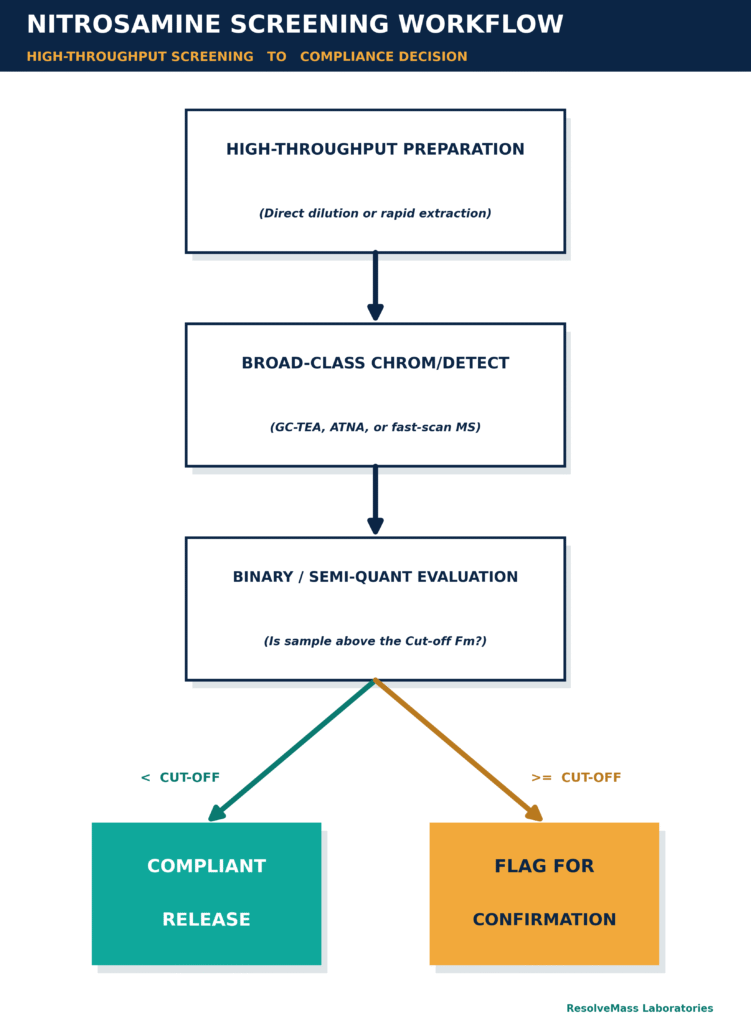
In screening applications, analytical speed and the ability to process a large number of samples are prioritized over absolute quantitative accuracy. One of the primary performance requirements is maintaining a false-negative rate (or β-error) below five percent, ensuring that potentially contaminated products do not unintentionally pass through the detection process.
Historically, nitrosamine screening has been performed using class-selective detection technologies, including the Thermal Energy Analyser (TEA) coupled with gas chromatography (GC-TEA) or Automated Total Nitrosamine Analyser (ATNA) platforms. The TEA detector selectively responds to chemiluminescent nitric oxide radicals (NO•) generated from pyrolyzed nitroso (N-NO) functional groups. This selective response produces a chromatographic profile with minimal interference from non-nitrosamine organic compounds, allowing efficient detection of nitrosamine-related impurities.
Alternatively, screening approaches may utilize rapid-scan mass spectrometry techniques, including high-performance liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) or gas chromatography-mass spectrometry (GC-MS/MS). These methods often incorporate simplified sample preparation procedures, such as direct dilution, enabling simultaneous screening of multiple analytes within a shorter analytical timeframe.
Since screening methods generally do not require extensive validation of compound-specific analytical parameters, such as absolute recovery evaluation or multi-point calibration curves across a wide concentration range, they provide an efficient and economical approach for initial assessment. These methods are particularly useful for screening raw materials, investigating excipient-related nitrite contamination, and confirming process-specific risk assumptions during early phases of drug development.
Unsure about your wider product portfolio obligations? Read our regulatory deep-dive on whether all drug products require a formal nitrosamine risk assessment.
Core Analytical Principles of Nitrosamine Confirmatory Methods
Nitrosamine confirmatory methods are highly specific and fully validated quantitative analytical procedures designed to accurately determine the chemical identity and concentration of individual nitrosamine impurities. These methods rely on advanced tandem mass spectrometry or high-resolution mass spectrometry techniques to provide the structural confirmation and quantitative accuracy necessary for regulatory submissions and demonstration of product safety at parts-per-billion levels.
The implementation of a confirmatory method becomes mandatory when an initial screening analysis produces a positive result or when a comprehensive risk assessment identifies a potential risk of nitrosamine formation or cross-contamination during manufacturing processes or long-term storage conditions.
Because regulatory acceptable daily intake (AI) limits for nitrosamines are extremely stringent, confirmatory methods must demonstrate exceptional sensitivity and achieve very low limits of quantitation (LOQ). For example, regulatory limits include 96 ng/day for N-nitrosodimethylamine (NDMA) and 26.5 ng/day for N-nitrosodiethylamine (NDEA). Therefore, confirmatory assays are generally designed to achieve an LOQ equivalent to ≤30% of the applicable AI limit.
To achieve the required sensitivity and selectivity, laboratories commonly utilize high-performance liquid chromatography coupled with triple quadrupole mass spectrometry (HPLC-MS/MS) operated in Multiple Reaction Monitoring (MRM) mode. High-Resolution Accurate Mass spectrometry (LC-HRMS), including advanced platforms such as Orbitrap systems, may also be employed for comprehensive structural confirmation and impurity characterization.
Confirmatory methods must undergo complete analytical validation according to ICH Q2(R2) requirements and applicable pharmacopeial guidelines. Validation studies must demonstrate appropriate performance characteristics, including linearity, accuracy, precision, specificity, sensitivity, and robustness within the relevant drug product matrix.
Ensure absolute compliance for your quantitative assays with our dedicated Nitrosamine Method Development and Validation Services.
Additionally, confirmatory assays require advanced chromatographic optimization to ensure complete separation of target nitrosamines from potential co-eluting compounds, including isobaric matrix components. These methods typically incorporate isotope-labeled internal standards, such as deuterated analogues including NDMA-d6, to compensate for variations caused by matrix effects, extraction recovery differences, and fluctuations in ionization efficiency.
By correcting analytical variability and minimizing the risk of inaccurate quantification, isotope-labeled standards enhance method reliability and ensure the generation of scientifically defensible data required for regulatory compliance and commercial batch release decisions.
Learn more about successfully meeting post-approval obligations through our overview of the Nitrosamine Batch Release Testing Requirements.
Systematic Comparison: Nitrosamine Screening vs Confirmatory Method
A direct comparison between a screening approach and a confirmatory assay demonstrates the strategic balance between analytical efficiency, laboratory throughput, and chromatographic specificity. This comparative assessment within the Nitrosamine Screening vs Confirmatory Method framework explains how different analytical strategies support various stages of the pharmaceutical development and manufacturing lifecycle.
Nitrosamine screening methods are primarily designed for rapid detection and risk identification, whereas confirmatory methods are developed for definitive identification, accurate quantification, and regulatory acceptance. Understanding these differences allows pharmaceutical manufacturers to select the appropriate analytical approach based on the intended purpose, regulatory expectations, and product-specific requirements.
The major operational and technical differences between screening and confirmatory methods are systematically summarized below:
| Technical Parameter | Screening Method | Confirmatory Method |
|---|---|---|
| Primary Objective | Class-level detection and identification of potentially contaminated or suspect batches. | Definitive identification and precise quantitation of specific target nitrosamine impurities. |
| Main Analytical Platforms | GC-TEA, ATNA, LC-MS/MS with simplified scanning approaches. | HPLC-MS/MS (QqQ), LC-HRMS (Orbitrap / QTOF). |
| Detection Principle | Chemiluminescence through N-NO cleavage or nominal mass transition detection. | Multiple Reaction Monitoring (MRM) or accurate mass extraction with high-resolution detection. |
| Validation Requirement | Limit test validation according to ICH Q2(R2). | Complete quantitative validation according to ICH Q2(R2) and USP requirements. |
| Quantitative Range | Semi-quantitative assessment performed against a defined cut-off limit. | Fully quantitative analysis generally covering 50% to 150% of the acceptable intake (AI) target concentration. |
| False Negative Rate (β-error) | Required to be less than 5% at the Screening Target Concentration (STC). | Extremely low and controlled through advanced chromatography and high-resolution mass spectrometric confirmation. |
| False Positive Rate (α-error) | Accepted during initial screening because positive results undergo confirmatory evaluation. | Strictly controlled through chromatographic separation and analytical specificity. |
| Internal Standards | Generally not required; external calibration approaches may be applied. | Isotope-labeled internal standards, including d3, d6, and 13C analogues, are required for accurate correction of analytical variability. |
| Typical Limit of Quantitation | Generally undefined or established at a broader Screening Target Concentration level. | ≤0.03 ppm for products with daily doses below 880 mg/day. |
| Matrix Suitability | Suitable for simpler matrices, raw materials, and excipient-related nitrite evaluation. | Required for complex drug products, highly concentrated APIs, and NDSRIs. |
| Regulatory Standing | Primarily used for internal risk evaluation and preliminary assessment; not suitable for batch release decisions. | Accepted for regulatory submissions, quality documentation, and commercial batch release. |
Regulatory Guidelines and Validation Protocols Under ICH Q2(R2) and USP
Global regulatory frameworks differentiate screening methods and confirmatory methods by classifying them as qualitative limit tests and quantitative impurity assays, respectively, under ICH Q2(R2) and relevant USP General Chapter guidelines. Validation of these analytical approaches requires compliance with specific performance criteria to ensure that trace-level nitrosamine measurements are scientifically reliable, reproducible, and suitable for regulatory inspection.
The United States Pharmacopeia (USP) establishes this analytical distinction through General Chapter requirements, which describe validated analytical procedures utilizing gas chromatography and liquid chromatography techniques coupled with mass spectrometry detection. Although these compendial procedures provide standardized analytical frameworks, laboratories often develop and validate customized equivalent methods to address specific drug product matrices, formulation characteristics, and targeted nitrosamine impurities.
To better understand the analytical procedures established within the USP compendial framework, the key parameters are summarized in the following table:
| USP Procedure | Analytical Platform | Targeted Nitrosamine Analytes | Sample Concentration & Compendial LOQ |
|---|---|---|---|
| Procedure 1 | LC-HRMS (Liquid Chromatography-High Resolution MS). | NDMA, NDEA, NEIPA, NDIPA, NMBA, NDBA, NMPA. | 20 mg/mL sample concentration; LOQ 0.05 μg/g (0.05 ppm). |
| Procedure 2 | GC-HS-MS/MS (Headspace Gas Chromatography-Triple Quad). | NDMA, NDEA, NEIPA, NDIPA. | 100 mg/mL sample concentration; LOQ 0.02 μg/g (0.02 ppm). |
| Procedure 3 | LC-MS/MS (Liquid Chromatography-Triple Quadrupole). | NDMA, NDEA, NEIPA, NDIPA, NMBA, NDBA. | 66.67 mg/mL sample concentration; LOQ 0.01 μg/g for NDEA and 0.02 μg/g for other analytes. |
| Procedure 4 | GC-MS/MS (Direct Injection Gas Chromatography-Triple Quad). | NDMA, NDEA, NEIPA, NDIPA, NDBA. | 100 mg/mL sample concentration; LOQ 0.005 μg/g (0.005 ppm). |
Learn how to properly establish internal safety thresholds by reviewing our comparison on Nitrosamine Alert Limits vs. Action Limits.
For a qualitative screening method validated as a limit test, the validation process under ICH Q2(R2) is comparatively simplified. The primary validation requirements include confirmation of specificity and detection capability. The method must demonstrate that it can reliably identify the targeted nitrosamine impurities at the predefined limit of interest without interference from the drug product matrix. Additionally, the experimentally determined limit of detection (LOD) must be scientifically established and verified to remain below the intended screening threshold.
In contrast, quantitative confirmatory methods require a much more comprehensive validation approach. These methods must undergo complete analytical performance characterization to demonstrate their suitability for accurate impurity quantification. The validated analytical range must extend from the established limit of quantitation (LOQ) through 150% of the concentration corresponding to the acceptable intake limit of the specific nitrosamine impurity.
The recommended performance criteria for validating a quantitative confirmatory nitrosamine method according to USP requirements and ICH Q2(R2) guidelines are summarized below:
| Validation Parameter | USP Compendial Requirement | Industry Best Practice Target |
|---|---|---|
| Linearity Range | 50%–150% of the specification limit. | LOQ to 150% of the target concentration. |
| Correlation Coefficient (r²) | ≥0.99 (Procedure 3 suitability standard). | ≥0.995 with acceptable back-calculated calibration values. |
| Accuracy (Percent Recovery) | 70%–130% recovery across the validated range. | 80%–120% recovery at medium and high spike levels. |
| Repeatability (Precision) | %RSD <25% for six replicate spiked samples (n=6). | %RSD ≤15% at target nitrosamine concentrations. |
| Intermediate Precision | Cumulative %RSD <30% for 12 determinations (n=12). | Cumulative %RSD ≤20% across different analysts and analytical days. |
| Limit of Quantitation (LOQ) | Verified according to maximum daily dose (MDD) and acceptable intake (AI) limits. | Signal-to-noise ratio (S/N) ≥10:1 with %RSD ≤20% precision. |
| Y-Intercept Deviation | ≤25% of the medium standard calibration response. | ≤10% of the target concentration standard response. |
The differences between screening and confirmatory methods highlight the importance of selecting an analytical strategy based on the intended regulatory purpose. Screening methods provide rapid and efficient risk identification, while confirmatory methods deliver the quantitative accuracy, specificity, and validation depth required for regulatory submissions and commercial product release.
Mechanical Techniques and Sample Preparation Protocols
Proper sample preparation is a critical analytical step that directly influences sensitivity, selectivity, and overall data quality during nitrosamine analysis. The primary objective of sample preparation is to effectively separate trace-level nitrosamine impurities from complex pharmaceutical matrices while reducing matrix-related interference, preventing analyte degradation, and minimizing the risk of artificial nitrosamine formation.
The physical handling and processing of solid and liquid pharmaceutical formulations involve several sensitive mechanical operations that require strict control under Good Manufacturing Practice (GMP) conditions. Even minor variations during sample manipulation can significantly affect extraction efficiency, recovery performance, and analytical reliability.
The contribution of different mechanical unit operations toward extraction efficiency and analytical consistency is summarized below:
Weighing:
Accurate measurement of reference standards and pharmaceutical samples is essential because even minor weighing deviations during low-milligram level preparation or spiking procedures can result in inaccurate concentration calculations. Analytical balances must undergo routine calibration, and weighing procedures should be carefully controlled to minimize variability introduced by operators.
Grinding:
Solid oral dosage forms, including coated and uncoated tablets, require effective pulverization into a fine and uniform powder to ensure representative sampling. Inadequate or inconsistent grinding may produce variable particle sizes, resulting in incomplete extraction, inconsistent analyte recovery, and increased analytical variation between samples or batches.
Sonication:
The application of ultrasonic energy during sample preparation improves extraction efficiency by disrupting the pharmaceutical matrix and facilitating the release of trace-level nitrosamine impurities into the extraction solvent. However, sonication conditions must be carefully optimized because excessive ultrasonic energy can generate localized temperature increases that may accelerate unwanted nitrosamine formation during preparation.
Vortexing and Shaking:
Mechanical mixing through vortexing or shaking promotes complete interaction between the sample matrix and extraction solvent. This improves phase-transfer efficiency and supports consistent recovery of both volatile and non-volatile nitrosamine impurities.
Centrifuging:
High-speed centrifugation separates extracted nitrosamine compounds present in the liquid supernatant from insoluble matrix components, including excipients such as microcrystalline cellulose, lactose, and magnesium stearate. This step improves sample cleanliness before chromatographic analysis.
Filtering:
The extracted supernatant is passed through a validated, chemically inert membrane filter to remove remaining particulate materials. Effective filtration protects chromatographic columns from contamination or blockage and ensures compatibility with sensitive mass spectrometry-based detection systems.
Chemical Mechanisms of False Positives and Artifactual Nitrosamine Formation
Artificial formation of nitrosamines during sample preparation represents a significant analytical challenge because it can generate false-positive results. This artifact occurs through the in-situ reaction between secondary or tertiary amines and residual nitrites under favorable chemical conditions, particularly acidic environments or elevated temperatures.
Preventing these unwanted reactions requires implementation of active chemical control strategies, including the use of nitrite scavengers, antioxidant additives, and optimized extraction conditions that maintain a controlled pH environment.
The fundamental mechanism of nitrosation involves the electrophilic reaction between a nitrosating species and an amine compound. Nitrous acid (HNO₂) is generated in situ when nitrite salts are exposed to acidic conditions (pH < 4.0). Following protonation, nitrous acid produces nitrous anhydride (N₂O₃), a highly reactive nitrosating agent that reacts with secondary amines to form stable N-nitrosamine compounds.
The reaction pathway can be represented as follows:
NO2− + H+ ⇌ HNO2
2HNO2 ⇌ N2O3 + H2O
R2NH + N2O3 → R2N-NO + HNO2
Common pharmaceutical excipients, including starch, lactose, and povidone, may contain trace levels of inorganic nitrites. When these excipients are combined with APIs containing secondary or tertiary amine functional groups, mechanical processing such as tablet pulverization may initiate nitrosation reactions directly within the extraction vessel.
Additionally, high-temperature sample preparation procedures or aggressive injection conditions may thermally degrade sensitive APIs, generating amine-containing intermediates that can subsequently undergo nitrosation reactions during analysis.
Deepen your technical knowledge of these chemical reactions by examining Nitrosamine Formation Pathways during API Synthesis.
To minimize and eliminate artificial nitrosamine formation, laboratories implement several scientifically controlled strategies:
Active pH Control:
Extraction solvents and mobile phases are adjusted to maintain neutral or mildly alkaline conditions (pH ≥ 7.0). This prevents conversion of nitrite ions into reactive nitrous acid, thereby reducing the possibility of nitrosation reactions.
Chemical Nitrite Scavengers:
Antioxidant-based scavengers, including ascorbic acid, sulfamic acid, and alpha-tocopherol, may be incorporated into extraction solvents. These compounds rapidly react with free nitrites and convert them into non-reactive nitric oxide species, interrupting the nitrosation pathway before amine compounds can form nitrosamines.
Chromatographic Probe Temperature Optimization:
Mass spectrometry ionization source and probe temperatures must be optimized to prevent thermal degradation of unstable nitrosamine compounds. Thermolabile nitrosamines are typically analyzed using lower probe temperature conditions, such as 150°C instead of 250°C, to minimize in-source fragmentation and artificial compound breakdown.
Managing Complex Nitrosamine Drug Substance-Related Impurities (NDSRIs)
Nitrosamine Drug Substance-Related Impurities (NDSRIs) represent one of the most challenging areas in nitrosamine analysis because they are chemically related to the active pharmaceutical ingredient (API). Unlike conventional nitrosamines, standardized screening approaches are often insufficient for NDSRI evaluation. Accurate quantitation requires development of product-specific confirmatory methods and, in many cases, custom synthesis of isotope-labeled internal standards to compensate for matrix effects and analytical challenges.
Examine the distinct differences and risk profiles by reading our comparison on NDSRIs vs. Simple Nitrosamines.
NDSRIs are structurally complex and generally non-volatile impurities formed when secondary amine functional groups within the API structure undergo nitrosation during pharmaceutical manufacturing, formulation processes, packaging interactions, or long-term storage. Examples of NDSRIs include N-nitroso-fluoxetine and N-nitroso-duloxetine.
Unlike small-molecule nitrosamines such as NDMA, which benefit from established analytical procedures and commercially available standards, each NDSRI requires a customized analytical strategy. Due to differences in molecular structure, physicochemical properties, and API-specific interactions, NDSRI methods must typically be developed individually using de novo analytical approaches.
The regulatory classification and analytical differences between small-molecule nitrosamines and complex NDSRIs are summarized below:
| Feature | Small-Molecule Nitrosamines | Nitrosamine Drug Substance-Related Impurities (NDSRIs) |
|---|---|---|
| Structural Complexity | Simple, volatile, low molecular weight organic compounds. | Complex, non-volatile compounds with structures unique to the parent API. |
| Formation Pathway | Associated with reagent carryover, contaminated solvents, or affected raw materials. | Generated through direct nitrosation of secondary or tertiary amine groups within the API structure. |
| Method Strategy | Standardized multi-analyte screening and confirmatory methods. | Customized, product-specific chromatographic methods developed through de novo method development. |
| Reference Standards | Commercially available Certified Reference Materials (CRMs). | Often unavailable commercially; requires custom organic synthesis. |
| Matrix Effects | Low to moderate, with relatively straightforward analytical separation. | Significant matrix challenges due to trace impurity detection within high-concentration API environments. |
| Acceptable Intake Limits | Historically established limits, such as NDMA at 96 ng/day. | Limits derived using the Carcinogenic Potency Categorization Approach (CPCA). |
Developing an effective NDSRI control strategy is highly complex because these impurities must be quantified at parts-per-billion levels while being present within a matrix containing extremely high concentrations of the parent API. The high API concentration can overwhelm the electrospray ionization (ESI) source of mass spectrometers, resulting in significant signal suppression and reduced analytical sensitivity.
Learn more about mitigating trace-level risks within complex matrices with our professional overview of Nitrosamine Testing for Highly Potent APIs.
Addressing these challenges requires advanced expertise in synthetic chemistry, mass spectrometry, and chromatographic method development. Analytical laboratories must optimize extraction conditions, separation mechanisms, and detection strategies to achieve reliable NDSRI measurement.
Contract research organizations (CROs) such as ResolveMass Laboratories Inc. address these analytical challenges through specialized in-house custom synthesis capabilities for producing high-purity, deuterated NDSRI reference standards. By combining these standards with advanced high-resolution Q-Exactive Orbitrap mass spectrometry platforms, laboratories can apply isotope dilution mass spectrometry approaches to achieve exceptionally low detection limits.
These advanced analytical strategies enable accurate NDSRI characterization and support compliance with stringent regulatory expectations, including the Carcinogenic Potency Categorization Approach (CPCA) frameworks established by regulatory authorities such as the FDA and Health Canada.
Strategic Implementation of Nitrosamine Testing in the Product Lifecycle
Integrating nitrosamine analysis throughout the pharmaceutical product lifecycle requires a structured, phased approach that aligns the analytical testing strategy with the specific stage of drug development and commercialization. Pharmaceutical manufacturers typically implement screening methods during early-stage raw material evaluation, supplier qualification, and risk assessment activities. As the product advances toward commercialization, these preliminary approaches are replaced with fully validated confirmatory methods to support regulatory submissions, commercial batch release, and long-term stability monitoring.
Product Lifecycle Nitrosamine Testing Strategy
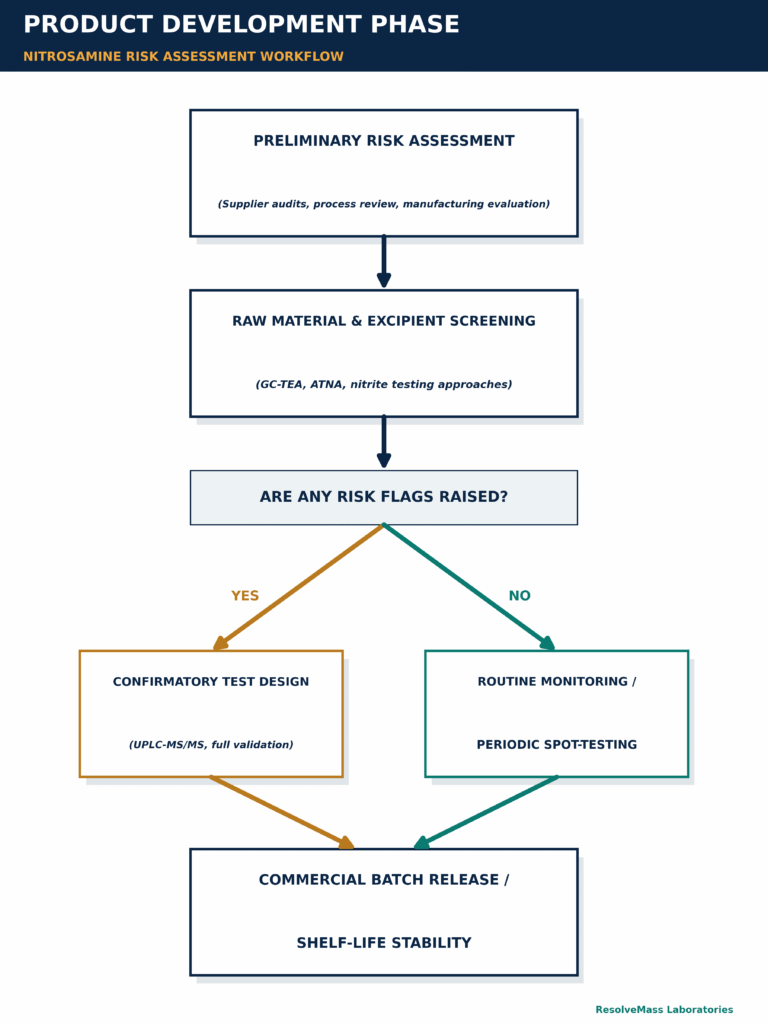
During the initial risk evaluation stage (Step 1), pharmaceutical manufacturers perform a detailed assessment of raw materials, solvent recovery systems, manufacturing pathways, and potential nitrosamine formation mechanisms. This evaluation includes reviewing supplier information, synthetic processes, formulation components, and potential sources of nitrite or amine contamination.
Plan your structural commercialization pathway accurately by viewing the standard Nitrosamine Testing Timeline.
At this stage, high-throughput screening techniques are commonly utilized to evaluate excipients for reactive nitrite precursors and to assess recovered solvents for potential amine-related contamination risks. Screening methods provide rapid identification of potential concerns while allowing manufacturers to efficiently evaluate large numbers of materials.
Ensure long-term quality alignment by considering the strategic advantages of outsourcing your nitrosamine testing program to a qualified CRO.
If no nitrosamine-related risks are identified and theoretical purge factor calculations demonstrate that potential impurities are effectively removed during manufacturing, companies may reduce or eliminate unnecessary routine commercial release testing requirements.
However, when a potential contamination risk is identified, or when preliminary screening produces a positive result, manufacturers must immediately proceed to Step 2: confirmatory testing. Confirmatory analytical methods are essential for obtaining definitive identification and quantitative measurement of nitrosamine impurities.
Confirmatory testing is particularly important for stability programs because nitrosamines may form gradually during storage due to factors such as packaging interactions, degradation pathways, or long-term exposure conditions. Therefore, validated confirmatory data is required to demonstrate product safety throughout the intended shelf life.
Additionally, confirmatory analytical results are necessary for regulatory documentation, including electronic Common Technical Document (eCTD) submissions associated with new marketing authorizations, post-approval changes, and alternative bioequivalence strategies.
By collaborating with a GMP-licensed analytical testing organization such as ResolveMass Laboratories Inc., pharmaceutical sponsors can ensure that their analytical datasets are scientifically justified, fully traceable, audit-ready, and aligned with Health Canada and FDA regulatory expectations.
Conclusion
Implementing a scientifically robust Nitrosamine Screening vs Confirmatory Method strategy is essential for ensuring patient safety and maintaining compliance with global regulatory requirements established by organizations such as the FDA, EMA, and Health Canada.
Working with a qualified analytical testing partner enables pharmaceutical companies to generate reliable data within a controlled quality system designed to support successful regulatory submissions and efficient product approvals across international markets.
Nitrosamine screening methods provide a rapid, economical approach for evaluating raw materials, excipients, and early-stage development samples. However, these approaches cannot replace the advanced mass spectrometric capabilities, analytical specificity, and quantitative accuracy required for final product release and regulatory compliance.
Selecting an analytical partner with extensive expertise in both synthetic chemistry and advanced mass spectrometry is critical for developing a comprehensive nitrosamine control strategy. A well-designed testing program helps minimize regulatory risks, prevent potential product recalls, and establish effective impurity management throughout the product lifecycle.
To learn more about analytical method validation, nitrosamine testing strategies, or to discuss your specific project requirements, contact the scientific experts at ResolveMass Laboratories Inc.
Frequently Asked Questions
Confirmatory testing becomes necessary when a risk assessment indicates a realistic possibility of nitrosamine contamination in an API or finished pharmaceutical product. It is also required when screening results indicate the presence of potential nitrosamines, during stability and shelf-life evaluations, and for regulatory submissions involving new approvals or post-approval manufacturing changes. These methods provide the definitive analytical evidence required for regulatory compliance.
Regulatory agencies establish acceptable daily intake (AI) limits for nitrosamines based on toxicological evaluations and carcinogenic risk assessments. For example, NDMA and NMBA have an AI limit of 96 ng/day, while compounds such as NDEA, NDIPA, and NEIPA have stricter limits of 26.5 ng/day. These limits define the maximum daily exposure considered acceptable for patient safety.
Nitrosamine concentration limits in pharmaceutical products are calculated by relating the acceptable daily intake (AI) value to the maximum daily dose (MDD) of the drug product. The calculation determines the allowable impurity concentration in parts-per-million (ppm) based on the patient’s maximum recommended daily exposure. This approach ensures that nitrosamine levels remain within established safety thresholds throughout product use.
Concentration Limit (ppm) = Acceptable Intake (ng) ÷ Maximum Daily Dose (mg)
Nitrosamines are considered highly potent mutagenic carcinogens, making accidental failure to identify contaminated products a significant patient safety concern. Maintaining a false-negative rate below 5% ensures that screening methods effectively identify the majority of potentially contaminated samples. This allows manufacturers to perform additional confirmatory testing before affected batches proceed further in the supply chain.
Artificial nitrosamine generation can occur when residual nitrites present in pharmaceutical excipients react with secondary or tertiary amine groups within the API during sample preparation. Factors such as acidic conditions, elevated temperatures, and high-energy processing can accelerate this reaction. Prevention strategies include controlling extraction pH, using high-quality solvents, optimizing analytical temperatures, and incorporating nitrite scavengers such as ascorbic acid to inhibit nitrosation.
GC-MS analysis may not be appropriate for thermally unstable pharmaceutical compounds because high injector temperatures can cause degradation of sensitive APIs. During this thermal breakdown, reactive amine intermediates may form and react with residual nitrites, leading to artificial generation of nitrosamines such as NDMA. In such situations, liquid chromatography coupled with mass spectrometry (LC-MS/MS) is preferred because it avoids thermal degradation during analysis.
Under USP requirements, confirmatory nitrosamine methods must undergo comprehensive analytical validation to demonstrate reliability and quantitative performance. Key criteria include specificity, linearity across 50%–150% of the specification limit, correlation coefficient (r²) of at least 0.99, acceptable recovery accuracy, precision requirements, and verification of the limit of quantitation (LOQ). These parameters ensure that the method can consistently detect and quantify nitrosamine impurities at regulatory limits.
Nitrosamine Drug Substance-Related Impurities (NDSRIs) are complex nitrosamine compounds formed from the chemical structure of the active pharmaceutical ingredient itself. They present significant analytical difficulties because each NDSRI is unique to a specific API, often lacks commercially available reference standards, and must be measured at extremely low concentrations. Additionally, the high concentration of the parent drug substance can create severe matrix effects and suppress mass spectrometric detection.
Reference:
- Manchuri, K. M., Shaik, M. A., Gopireddy, V. S. R., Sultana, N., & Gogineni, S. (2024). Analytical methodologies to detect N-nitrosamine impurities in active pharmaceutical ingredients, drug products and other matrices. Chemical Research in Toxicology, 37(9), 1456–1483. https://doi.org/10.1021/acs.chemrestox.4c00234
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (1995). Validation of analytical procedures: Text and methodology (ICH Q2(R1)) (Step 5). European Medicines Agency. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-q2r1-validation-analytical-procedures-text-methodology-step-5-first-version_en.pdf
- Nichani, K., Uhlig, S., Stoyke, M., Kemmlein, S., Ulberth, F., Haase, I., & Walch, S. G. (2023). Essential terminology and considerations for validation of non-targeted methods. Food Chemistry: X, 17, 100538. https://doi.org/10.1016/j.fochx.2022.100538
- Sudheer, M. (2025, November 26). What are the acceptable validation limits for nitrosamine impurity testing, and which regulatory references define them? ResearchGate. https://www.researchgate.net/post/What_Are_the_Acceptable_Validation_Limits_for_Nitrosamine_Impurity_Testing_and_Which_Regulatory_References_Define_Them
- United States Pharmacopeia. (2023). <1469> Nitrosamine impurities: Highlights of general chapter. United States Pharmacopeia. https://www.usp.org/sites/default/files/usp/document/stakeholder-forum/pnp/highlights-of-1469-nitrosamine-impurities.pdf
- U.S. Food and Drug Administration. (2024). CDER nitrosamine impurity acceptable intake limits. Center for Drug Evaluation and Research, U.S. Food and Drug Administration. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cder-nitrosamine-impurity-acceptable-intake-limits
- Health Canada. (2024). Nitrosamine impurities in medications: Guidance for industry. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities/medications-guidance.html

