
Maintaining a predictable Nitrosamine Testing Timeline has become a critical operational requirement for pharmaceutical manufacturers working to meet stringent compliance expectations established by the Food and Drug Administration (FDA), Health Canada, and the European Medicines Agency (EMA). Since nitrosamines are classified as potent mutagenic carcinogens, regulatory agencies require highly sensitive and fully validated analytical procedures to verify that trace impurity concentrations remain within acceptable limits. However, the time required to complete nitrosamine studies is not standardized across all products. Instead, the overall timeline depends on factors such as the structural complexity of the analyte, the characteristics of the drug matrix, and the specific type of study being performed.
Article Summary:
- Nitrosamine testing timelines typically range from 2 to 6 weeks, depending on the study scope, product complexity, analytical requirements, and regulatory expectations.
- A complete testing program includes multiple stages such as risk assessment, method development, custom impurity synthesis, method validation, confirmatory testing, and final quality review, each contributing to the overall project duration.
- Initial risk assessments can often be completed within a few days, while laboratory screening helps identify potential nitrosamine contamination and determines whether further quantitative testing is required.
- Developing and validating GMP-compliant analytical methods is one of the most time-intensive steps, requiring extensive optimization to accurately detect nitrosamines at extremely low concentrations and satisfy ICH regulatory requirements.
- Custom synthesis and characterization of Nitrosamine Drug Substance-Related Impurities (NDSRIs) frequently represent the largest bottleneck because specialized reference standards must be created, purified, and fully characterized before validation can proceed.
- Analytical technology selection significantly affects turnaround times. Advanced platforms such as Headspace GC-MS/MS and High-Resolution Mass Spectrometry (HRMS) improve efficiency, reduce false positives, and accelerate investigations and routine testing workflows.
- Project delays commonly arise from matrix interferences, challenging sample preparation, artefactual nitrosamine formation, and limited availability of reference standards. Early planning, parallel workflows, and integrated analytical capabilities can substantially shorten testing timelines and improve regulatory readiness.
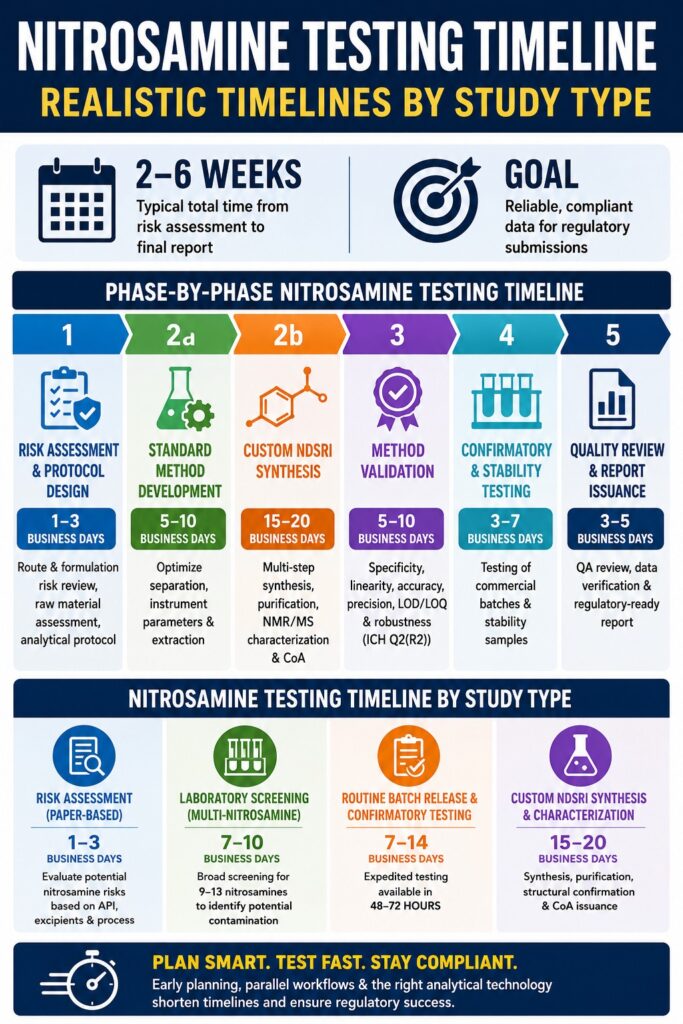
Phase-by-Phase Breakdown of the Nitrosamine Testing Timeline
A typical nitrosamine testing and validation program generally requires between 2 and 6 weeks to progress from the initial chemical risk assessment stage through final regulatory report issuance. This structured workflow enables analytical chemistry teams to identify and eliminate matrix-related interferences, establish dependable quantitation limits, and generate scientifically defensible data before regulatory submissions are prepared.
The overall progression of a nitrosamine testing project is driven by a series of sequential analytical milestones. This is especially important when evaluating non-standard active pharmaceutical ingredients (APIs) or novel drug substance-related impurities. The phase-by-phase Nitrosamine Testing Timeline is summarized below:
| Phase | Milestone | Estimated Duration | Core Deliverables & Operational Focus |
|---|---|---|---|
| Phase 1 | Risk Assessment & Protocol Design | 1–3 Business Days | Compilation of synthetic route evaluations, formulation risk assessments, raw material assessments, and formal analytical protocols. |
| Phase 2a | Standard Method Development | 5–10 Business Days | Optimization of chromatographic separation (LC or GC), mass spectrometer source tuning, and sample extraction parameters. |
| Phase 2b | Custom NDSRI Synthesis | 15–20 Business Days | Multi-step organic synthesis, chromatographic purification, NMR and mass spectral characterization, and Certificate of Analysis (CoA) preparation. |
| Phase 3 | Formal Method Validation | 5–10 Business Days | Assessment of specificity, linearity, accuracy, precision, limits of detection and quantitation, and robustness according to ICH Q2(R2) requirements. |
| Phase 4 | Confirmatory & Stability Testing | 3–7 Business Days | Extraction procedures and ultra-trace quantitative analysis of representative commercial and stability batches. |
| Phase 5 | Quality Review & Report Issuance | 3–5 Business Days | Comprehensive Quality Assurance (QA) review, raw data verification, and preparation of regulatory-ready documentation. |
Estimating the Nitrosamine Testing Timeline by Study Type
The Nitrosamine Testing Timeline varies significantly depending on the type of study being conducted. Paper-based evaluations can often be completed within 1 to 3 business days, whereas custom impurity synthesis projects combined with full GMP validation activities may require up to 4 weeks. Aligning project schedules with realistic testing timelines helps manufacturers satisfy rolling submission deadlines while avoiding disruptions to commercial supply.
Timelines for Risk Assessment and Initial Screening
A paper-based risk assessment can generally be completed within 1 to 3 business days, while laboratory-based multi-nitrosamine screening studies typically require 7 to 10 business days. These preliminary activities are designed to determine whether a formulation contains chemical vulnerabilities that justify formal quantitative investigation.
The initial risk assessment involves a comprehensive evaluation of factors that may contribute to nitrosamine formation. This includes reviewing the presence of secondary or tertiary amines within the API, identifying nitrite-containing excipients, and assessing the potential for contamination originating from solvents, reagents, or raw materials. The outcome of this assessment provides the scientific rationale for subsequent testing activities.
When chemical risks are identified, laboratories proceed with Phase 1 screening using broad-spectrum multi-nitrosamine panels. These analytical panels commonly monitor between 9 and 13 well-known small-molecule nitrosamines, including NDMA, NDEA, and NDBA. Because these screening studies do not require complete validation for every analyte-matrix combination, results can be generated relatively quickly, allowing manufacturers to identify potential contamination before investing in more comprehensive testing programs.
Ensure your regulatory strategy is sound from day one. Review how to structure your paperwork effectively by visiting the Nitrosamine Risk Assessment for ANDA Submission Guide.
Impact of Method Development on the Nitrosamine Testing Timeline
The development and validation of a custom quantitative analytical method under GMP conditions generally requires between 10 and 20 business days. This timeframe is necessary to optimize sample preparation procedures and instrument performance to reliably detect target analytes at sub-parts-per-billion concentrations.
Method development begins with establishing the required Limit of Quantitation (LOQ). Regulatory expectations typically require the LOQ to be at or below 30% of the impurity’s acceptable intake (AI) limit, although many laboratories target an LOQ ≤ 10% of the AI to maximize regulatory confidence. During development, analytical scientists optimize several critical parameters, including HPLC column selection, mobile phase composition and gradients, gas chromatography temperature programs, and tandem mass spectrometry (MS/MS) precursor-to-product ion transitions.
Following optimization, the analytical method must undergo a formal validation process in accordance with ICH Q2(R2) and ICH M7(R2) requirements. This extensive validation confirms that the procedure is accurate, precise, specific, reproducible, and sufficiently robust for routine commercial use and long-term regulatory compliance.
Learn more about establishing compliant parameters for your analytical workflows by checking out the Nitrosamine Specification Setting Technical Protocol.
Turnaround Times for Routine Batch Release and Confirmatory Testing
Routine batch release testing is generally completed within 7 to 14 business days. However, many laboratories also offer expedited testing programs capable of delivering compliant results within 48 to 72 hours. These turnaround times are designed to minimize product hold periods while ensuring that batches exceeding established safety limits do not enter the market.
Confirmatory testing is initiated when risk assessments identify a plausible nitrosamine formation pathway. To satisfy regulatory expectations, testing must be performed on a representative selection of newly manufactured commercial batches as well as retained stability samples. This approach provides insight into impurity behavior throughout the product’s intended shelf life.
If confirmatory testing demonstrates that nitrosamine concentrations consistently remain below 10% of the established AI limit, routine release specifications are generally not required. Conversely, when impurity levels exceed the 10% threshold, formal release specifications must be implemented, resulting in a mandatory testing program as outlined below:
| Impurity Concentration (Relative to AI) | Testing Requirement & Frequency | Required Regulatory Submission |
| < 10% AI | Routine testing generally not required; periodic audit testing may be appropriate. | Results documented within the product risk assessment dossier. |
| 10%–30% AI | Routine release testing required; skip-testing may be considered after sufficient validation. | Formal release specifications established within the registered dossier. |
| 30%–100% AI | Mandatory batch-by-batch testing for all commercial lots. | Dossier updated to include mandatory release specifications. |
| ≥ 100% AI | Batch rejection and manufacturing hold. | Immediate regulatory notification and market recall if product has been distributed. |
Understand the mandatory compliance criteria for commercial lots by visiting the Nitrosamine Batch Release Testing Requirement Breakdown.
Timeline for Custom NDSRI Synthesis and Reference Standard Characterization
The synthesis and characterization of novel Nitrosamine Drug Substance-Related Impurities (NDSRIs) generally require between 15 and 20 business days. This stage frequently represents one of the most significant bottlenecks within the Nitrosamine Testing Timeline because commercially available reference standards rarely exist for custom NDSRIs.
NDSRIs are structurally related to the API and may form when secondary or tertiary amines present within the drug molecule react with trace nitrites found in the formulation. Because these impurities are highly compound-specific, certified reference standards must be synthesized to establish chromatographic retention behavior and optimize mass spectrometric detection parameters.
The synthesis process typically involves multiple organic reaction steps followed by advanced purification procedures. Structural identity and purity are subsequently confirmed using nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS). After characterization is completed, the reference standard is issued with a GMP-compliant Certificate of Analysis (CoA), enabling subsequent method validation activities.
Explore the structural factors driving NDSRI risk evaluation on the Nitrosamine CPCA Approach for NDSRIs Analysis.
Analytical Technologies Influencing the Nitrosamine Testing Timeline
The selection of analytical instrumentation directly affects both the speed and reliability of the Nitrosamine Testing Timeline. In many cases, headspace GC-MS/MS provides faster routine testing workflows than more complex liquid chromatographic extraction methods. Selecting the most appropriate analytical platform based on the physical and chemical characteristics of the target impurity helps reduce instrument downtime and maintain predictable study schedules.
Direct Injection vs Headspace GC-MS/MS Timelines
Headspace GC-MS/MS minimizes maintenance-related downtime by preventing non-volatile matrix components from entering the chromatographic system. Direct injection approaches, while often offering enhanced sensitivity for heavier semi-volatile compounds, typically require more frequent cleaning and maintenance. The choice between these approaches depends largely on analyte volatility.
Direct injection methods involve dissolving the complete sample matrix and introducing the resulting extract directly into the GC or LC system. While this approach can provide excellent recovery for semi-volatile and non-volatile nitrosamines, it also introduces excipients, polymers, and concentrated API components onto the chromatographic column. Over time, this accumulation can result in signal suppression, peak distortion, and instrument contamination, ultimately requiring maintenance and column replacement.
Headspace GC-MS/MS operates differently by relying on gas-phase partitioning. Samples are heated within sealed vials, allowing volatile analytes to migrate into the vapor phase. Only the vapor fraction is introduced into the analytical system. As a result, non-volatile excipients and API components remain in the vial, preserving instrument cleanliness and supporting high-throughput testing with minimal downtime.
| Feature | Direct Injection GC/LC-MS/MS | Headspace GC-MS/MS |
| Mechanism | Introduces liquid sample extract directly into the instrument. | Introduces vapor-phase analytes equilibrated above the sample matrix. |
| Analyte Suitability | Volatile, semi-volatile, and non-volatile nitrosamines. | Volatile nitrosamines such as NDMA, NDEA, and NDIPA. |
| Matrix Exposure | High exposure; column and MS source susceptible to contamination. | Minimal exposure; non-volatile matrix components remain in the vial. |
| Throughput & Speed | Moderate throughput due to sample clean-up and maintenance requirements. | High throughput with extensive automation and reduced downtime. |
| Limit of Quantitation | Highly sensitive for heavier semi-volatile compounds. | Exceptional sensitivity for highly volatile analytes. |
High-Resolution Mass Spectrometry (HRMS) Solutions
High-Resolution Mass Spectrometry (HRMS) significantly accelerates troubleshooting activities and out-of-specification (OOS) investigations by using exact mass measurements to distinguish target nitrosamines from co-eluting isobaric compounds. This capability reduces the risk of false-positive findings that can interrupt manufacturing operations for extended periods.
Traditional triple-quadrupole systems operate at unit mass resolution and may generate false positives when matrix-derived fragments share nominal mass transitions with target nitrosamines. HRMS platforms, including Orbitrap and Time-of-Flight (Q-TOF) systems, provide ultra-high mass resolution and mass accuracy below 3 ppm, allowing co-eluting compounds to be distinguished based on exact molecular composition.
In addition, HRMS systems employ Data-Independent Acquisition (DIA), which captures comprehensive full-scan accurate-mass datasets. This feature allows laboratories to retrospectively evaluate previously acquired data for newly regulated nitrosamines without rerunning physical samples, potentially saving weeks of laboratory effort.
Discover practical applications of advanced mass spectrometry platforms by reading the NDMA Root Cause Investigation Case Study.
Critical Regulatory Milestones and Thresholds Dictating Timelines
The Nitrosamine Testing Timeline is strongly influenced by toxicological thresholds and implementation deadlines established by international regulatory authorities. When analytical results exceed specified limits, manufacturers may be required to transition rapidly from periodic monitoring to mandatory batch-release testing.
Under current ICH M7(R2) and FDA guidance, acceptable intake limits are calculated using compound-specific carcinogenicity data. When direct toxicological information is available, the acceptable intake (AI) can be calculated from the TD{50} value:
\text{AI} = \frac{TD_{50} \times 50\text{ kg}}{50,000}
Where 50 kg represents the assumed average body weight and 50,000 is the safety factor applied to maintain a theoretical lifetime cancer risk below 1 in 100,000.
When direct TD{50} data are unavailable, the Carcinogenic Potency Categorization Approach (CPCA) is used to classify NDSRIs according to structural characteristics, electronic properties, and metabolic activation potential. The categories and corresponding AI limits are outlined below:
| CPCA Category | Structural Characteristics & Reactivity Indicators | Daily AI Limit (ng/day) | 10% Testing Threshold Trigger |
| Category 1 | High carcinogenic activation potential with no deactivating groups. | 26.5 | 2.65 ng/day |
| Category 2 | Intermediate structural characteristics with moderate metabolic activation potential. | 100 | 10.0 ng/day |
| Category 3 | Presence of moderate deactivating features or steric hindrance. | 400 | 40.0 ng/day |
| Category 4 | Significant deactivating groups such as electron-withdrawing substituents. | 1500 | 150.0 ng/day |
| Category 5 | Highly deactivating structural features or severe steric hindrance. | 1500 | 150.0 ng/day |
For a detailed breakdown of toxicity classification rules and calculation strategies, see the Nitrosamine AI Limit and CPCA Regulatory Guide.
Operational Challenges That Delay the Nitrosamine Testing Timeline
Unexpected delays ranging from several days to multiple weeks are commonly caused by matrix interferences, sample preparation difficulties, and reference standard procurement issues. Addressing these challenges during protocol development significantly reduces the likelihood of project delays.
Matrix Swelling and Interference: Hydrophilic polymers such as hydroxypropyl methylcellulose (HPMC), frequently used in extended-release formulations, can form highly viscous gels when exposed to extraction solvents. These gels may trap nitrosamines and result in poor recoveries and inconsistent validation outcomes. Resolving these issues often requires advanced clean-up procedures such as Solid-Phase Extraction (SPE) or Liquid-Liquid Extraction (LLE), which may add 3 to 5 business days to method development.
Artefactual Nitrosamine Formation: During sample preparation, trace amounts of secondary or tertiary amines may react with ambient nitrites under acidic conditions, generating artificial nitrosamines within the sample vial. Such reactions can lead to false-positive results. Laboratories frequently address this challenge by incorporating secondary amine scavengers, including nucleophilic quenchers such as cysteine or ascorbate, or radical scavengers such as BHT and tocopherols, which selectively intercept nitrosyl cations ($NO^+$) before reaction with amines can occur.
Reference Standard Chokepoints: Obtaining certified reference standards for custom NDSRIs remains a significant operational challenge. Because many NDSRIs are not commercially available, manufacturers that delay confirmatory testing or rely solely on theoretical risk assessments often encounter regulatory obstacles, forcing custom synthesis projects to begin late in the compliance timeline.
Learn how matrix modifications and stabilization ingredients mitigate false-positive risks in the Analysis of Secondary Amine Scavengers for Nitrosamines.
Strategic Recommendations for Accelerating the Nitrosamine Testing Timeline
Reducing the overall Nitrosamine Testing Timeline requires implementation of parallel workflows that integrate risk assessments, custom synthesis activities, and method development at the earliest possible stage of product development. Collaborating with contract research laboratories that provide both custom synthesis capabilities and high-resolution mass spectrometry under one roof can eliminate many technology-transfer delays.
Rather than performing development activities sequentially, manufacturers can initiate custom reference standard synthesis (Phase 2b) concurrently with analytical method development (Phase 2a). In addition, establishing highly sensitive methods with LOQs substantially below the 10% AI threshold enables the generation of robust multi-batch datasets that may support Option 4 waivers under ICH M7.
An Option 4 waiver demonstrates that the manufacturing process possesses sufficient purge capability to consistently eliminate mutagenic impurities. As a result, ongoing routine batch-release testing may no longer be necessary, reducing future compliance burdens while strengthening supply chain reliability.
Learn how to mathematically establish your process’s safety profile by reading the Technical Guide on Nitrosamine Purge Factor Calculations.
Conclusion
Successfully reducing the Nitrosamine Testing Timeline requires technical foresight, disciplined project planning, and early alignment with evolving international regulatory expectations. Effectively managing the requirements associated with custom NDSRI synthesis and method validation under ICH Q2(R2) supports long-term commercial success while helping protect public health from carcinogenic impurities.
By establishing realistic project timelines and selecting appropriate analytical technologies, pharmaceutical manufacturers can move beyond reactive troubleshooting and adopt a proactive compliance strategy. For direct assistance with custom method development, GMP validation, and expedited testing programs, organizations can contact the expert team at Resolve Mass Laboratories through the contact page.
Frequently Asked Questions
Routine testing becomes mandatory when confirmatory studies show that a nitrosamine impurity exceeds 10% of its established Acceptable Intake (AI) limit. At that point, regulatory authorities generally expect the impurity to be incorporated into the product’s release specifications. Manufacturers must then implement a formal testing strategy for commercial batches unless an alternative, scientifically justified control approach has been accepted by regulators.
Regulatory guidance requires the Limit of Quantitation (LOQ) to be no greater than 30% of the applicable acceptable intake limit for the target impurity. However, many laboratories aim for significantly greater sensitivity by developing methods with LOQs at or below 10% of the AI limit. Achieving lower LOQs strengthens data quality, improves regulatory confidence, and may support future requests for reduced testing requirements.
The 30% AI threshold serves as an important regulatory decision point when establishing impurity control strategies. Nitrosamine levels below this threshold may allow manufacturers to justify reduced monitoring approaches, depending on supporting data. Once concentrations rise above 30% of the acceptable intake limit, regulators generally require routine batch-by-batch testing to ensure consistent product safety and compliance.
Small-molecule nitrosamines, such as NDMA and NDEA, are low-molecular-weight contaminants that are not structurally related to the active pharmaceutical ingredient. In contrast, Nitrosamine Drug Substance-Related Impurities (NDSRIs) are chemically linked to the API and arise from reactions involving amine-containing drug molecules and trace nitrites. Because NDSRIs are drug-specific, their identification and control often require customized analytical approaches.
When direct carcinogenicity information is unavailable, regulators rely on the Carcinogenic Potency Categorization Approach (CPCA) to establish an acceptable intake limit. This framework evaluates structural characteristics, metabolic activation potential, and the presence of deactivating functional groups. Based on these factors, the impurity is assigned to a potency category with a corresponding AI limit ranging from 26.5 ng/day to 1500 ng/day.
The Maximum Daily Dose (MDD) has a direct impact on the concentration limits that laboratories must measure. Since acceptable intake values are expressed as a daily mass exposure, the allowable concentration in the product is calculated relative to the total daily dose. Products with higher MDDs typically require lower detection limits, making highly sensitive techniques such as LC-MS/MS essential for compliance.
Under accelerated testing programs, many GMP-compliant laboratories can provide routine release or confirmatory nitrosamine testing results within 48 to 72 hours. These expedited services are often used when manufacturers face urgent production schedules, supply chain constraints, or regulatory deadlines. Although turnaround times are shortened, laboratories must still maintain full compliance with quality and documentation requirements.
Secondary amine scavengers are specialized chemical additives used during sample preparation to prevent artificial nitrosamine formation. Compounds such as cysteine, ascorbate, BHT, and tocopherols react with nitrosating species before they can interact with amine-containing drug substances. Their use helps ensure that measured nitrosamine levels accurately reflect the sample itself rather than artifacts generated during extraction and analysis.
Reference:
- U.S. Food and Drug Administration. (2024, September). Control of nitrosamine impurities in human drugs: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/control-nitrosamine-impurities-human-drugs
- United States Pharmacopeia. (2021). General chapter <1469> Nitrosamine impurities. USP–NF. United States Pharmacopeia. https://doi.org/10.31003/USPNF_M15715_02_01
- United States Pharmacopeia (USP). <1469> Nitrosamines in Drug Products. USP-NF. https://www.usp.org
- Health Canada. (2023, October). Guidance on Nitrosamine Impurities in Medications. Government of Canada. https://www.canada.ca/content/dam/hc-sc/documents/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities/medications-guidance/guidance-nitrosamine%20impurities-medications.pdf

