
Introduction:
Pharmaceutical manufacturers function under rigorous oversight from major global regulatory agencies, including the United States Food and Drug Administration (FDA), the European Medicines Agency (EMA), and Health Canada, to monitor and control the presence of mutagenic impurities in pharmaceutical products. Following the large-scale recalls involving sartan-containing antihypertensive medications, metformin products, and ranitidine formulations, regulatory bodies introduced extremely stringent permissible daily intake thresholds for carcinogenic N-nitrosamines. Achieving compliance with these demanding regulatory expectations requires advanced scientific expertise, highly specialized analytical methodologies, and sophisticated mass spectrometry instrumentation. As a result, outsourcing nitrosamine testing to a CRO has become the preferred operational strategy for pharmaceutical companies seeking reliable analytical data, regulatory compliance, and enhanced patient safety.
Executive Summary:
- Regulatory agencies such as the FDA, EMA, and Health Canada now enforce extremely strict limits for nitrosamine impurities following major pharmaceutical product recalls involving ranitidine, metformin, and sartan drugs.
- Outsourcing nitrosamine testing to a CRO has become a preferred strategy because it provides access to advanced mass spectrometry platforms, regulatory expertise, and validated analytical workflows.
- A typical nitrosamine testing program includes multiple stages such as risk assessment, custom analytical method development, formal method validation, confirmatory testing, and preparation of regulatory-compliant documentation.
- Risk assessments evaluate APIs, raw materials, solvents, intermediates, packaging systems, and storage conditions to identify possible nitrosamine formation pathways.
- CROs commonly use high-sensitivity analytical platforms such as LC-MS/MS, GC-MS/MS, and high-resolution Orbitrap mass spectrometry to detect ultra-trace nitrosamine impurities at parts-per-billion or parts-per-trillion levels.
- Method validation must comply with ICH Q2(R2) and M7(R2) guidelines, including demonstration of specificity, precision, accuracy, robustness, and reliable LOQ performance in complex pharmaceutical matrices.
- Project timelines generally range from 2 to 6 weeks, depending on the complexity of the study, while costs may vary from approximately $1,000 for limited verification work to more than $35,000 for full validation and custom NDSRI synthesis projects.
- Selecting the right CRO requires careful evaluation of regulatory inspection history, analytical expertise, data integrity practices, instrumentation capability, and experience handling nitrosamine-related compliance programs.
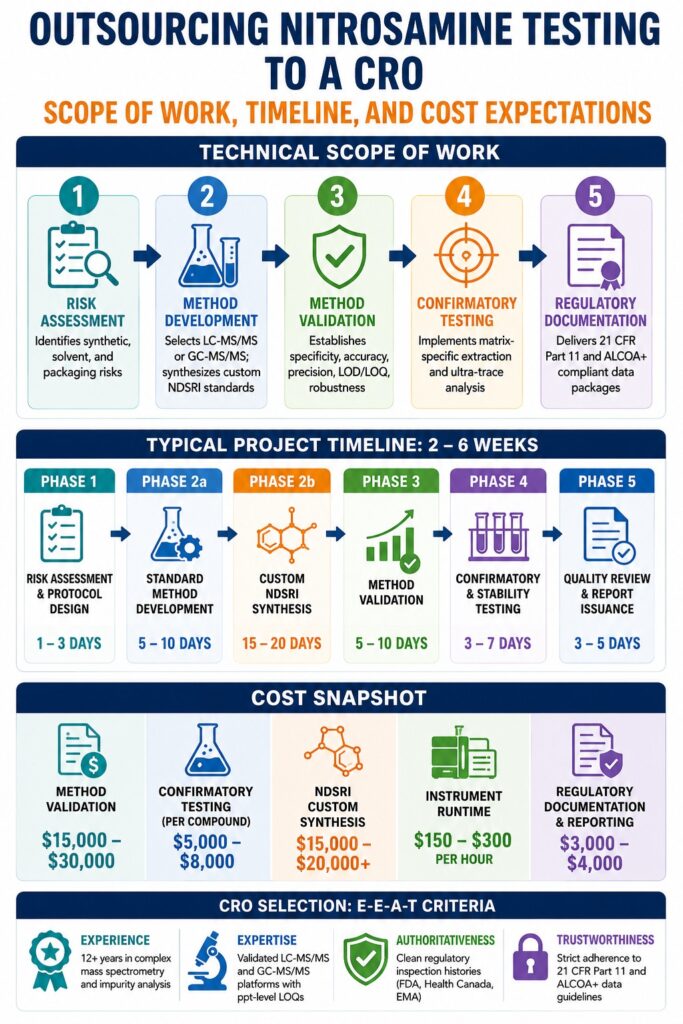
Navigating the Technical Scope of Work When Outsourcing Nitrosamine Testing to a CRO
The technical scope involved in outsourcing nitrosamine testing to a CRO follows a structured, multi-stage framework that includes comprehensive risk assessments, customized analytical method development, formal validation according to ICH guidelines, confirmatory testing, and regulatory documentation preparation. This systematic lifecycle ensures that all potential mutagenic impurity pathways are identified, evaluated, and controlled appropriately.
+------------------------------------------------------------------------------------------+
| TECHNICAL SCOPE OF WORK |
+------------------------------------------------------------------------------------------+
| 1. RISK ASSESSMENT ==> Identifies synthetic, solvent, and packaging risks |
| │ |
| ▼ |
| 2. METHOD DEV. ==> Selects LC-MS/MS or GC-MS/MS; synthesizes custom NDSRI standards|
| │ |
| ▼ |
| 3. METHOD VAL. ==> Establishes specificity, accuracy, precision, LOD/LOQ, robustness|
| │ |
| ▼ |
| 4. CONFIRMATORY ==> Implements matrix-specific extraction and ultra-trace analysis |
| │ |
| ▼ |
| 5. REGULATORY ==> Delivers 21 CFR Part 11 and ALCOA+ compliant data packages |
+------------------------------------------------------------------------------------------+Stage 1: Nitrosamine Risk Assessments (NRA)
Sponsors are required to assess raw materials, active pharmaceutical ingredient (API) synthesis pathways, manufacturing solvents, process intermediates, and storage environments. Contract research organizations examine the possibility of secondary or tertiary amines reacting with nitrosating agents, such as sodium nitrite, particularly under acidic conditions that may facilitate nitrosamine formation.
The risk assessment process also extends to packaging systems. Research has demonstrated that blister packaging materials, elastomeric closures, and plastic storage containers may contribute to nitrosamine generation. Trace-level amines, reactive packaging films, and chemically active printing lacquers may interact under elevated temperature and high-humidity storage conditions, increasing the likelihood of impurity formation over time.
Explore specialized analytical testing strategies for container-closure systems and foil migration concerns at the Packaging Leachables and Nitrosamine E&L Guide.
Stage 2: Custom Analytical Method Development
When the risk assessment identifies potential chemical vulnerabilities, the CRO must design and optimize a targeted analytical method capable of accurately detecting the identified nitrosamine species. The physicochemical properties of the target nitrosamine determine the most suitable analytical platform. Polar and non-volatile nitrosamines are typically analyzed using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS), while volatile nitrosamines are generally evaluated using Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS) or Headspace GC-MS techniques.
For Nitrosamine Drug Substance-Related Impurities (NDSRIs) that do not have commercially available reference standards, the contract laboratory must conduct custom organic synthesis. API-derived precursor molecules are reacted with nitrosating agents under controlled laboratory conditions to generate appropriate analytical reference materials. These synthesized standards subsequently undergo purification and structural confirmation through high-field Nuclear Magnetic Resonance (NMR) spectroscopy and High-Resolution Mass Spectrometry (HRMS).
Evaluate whether vapor-phase extraction or direct liquid introduction fits your specific API matrix at the Direct Injection vs. Headspace Techniques for Nitrosamines Comparison.
Crucial Analytical Stages When Outsourcing Nitrosamine Testing to a CRO
The analytical workflow progresses through systematic impurity screening, synthesis of custom reference materials, and high-sensitivity chromatographic separation techniques designed to isolate ultra-trace contaminants from complex APIs and excipient matrices. These highly specialized stages are essential for establishing analytical workflows capable of reliable parts-per-billion detection.
STAGED METHOD DEVELOPMENT AND QUANTIFICATION FLOW
──► Solid, Liquid, or Tablet Form
│
▼
──► Centrifugation / Matrix Cleanup
│
▼
──► Addition of Stable Isotope Labels
│
▼
──► UHPLC (C18 Column) or Capillary GC
│
▼
──► Triple Quadrupole MRM Transitions To achieve highly accurate quantification, analytical chemists must optimize multiple fragmentation pathways, ion source parameters, collision energies, and Multiple Reaction Monitoring (MRM) transitions. In formulated drug products, the CRO also optimizes extraction procedures while carefully evaluating excipient interference and its impact on analyte recovery. When acceptable recovery thresholds cannot be consistently achieved, stable isotope-labeled internal standards are introduced to compensate for matrix effects and signal suppression.
In addition, forced degradation studies are conducted under acidic, alkaline, oxidative, and thermal stress conditions to characterize degradation pathways and support scientifically justified shelf-life recommendations.
Understand how to structure your long-term stability testing protocols to monitor potential impurity formation over time by reviewing the Nitrosamine Testing in Stability Studies Framework.
Technical Method Validation Requirements and Analytical Platforms
Validation of analytical methods for nitrosamine impurities must comply fully with ICH Q2(R2) and M7(R2) guidelines to establish method specificity, accuracy, precision, robustness, and reproducibility across defined concentration ranges. Advanced analytical laboratories rely heavily on tandem mass spectrometry and high-resolution mass spectrometry platforms to achieve limits of quantification as low as 0.1\text{ ng/mL}.
To satisfy the expectations of international health authorities, analytical methods must undergo comprehensive validation demonstrating that ultra-trace nitrosamine residues can be consistently and accurately quantified in the presence of highly complex pharmaceutical matrices. Validation parameters must be thoroughly documented because incomplete validations, inadequate robustness testing, or missing system suitability requirements can lead to significant regulatory concerns or outright submission rejection.
Analytical Platforms and Target Validation Specifications
| Analytical Platform | Target Nitrosamine Class | Target Limits (LOD / LOQ) | Validation Recovery Limits | Key Technical Advantage |
|---|---|---|---|---|
| Triple Quadrupole LC-MS/MS | Polar, non-volatile nitrosamines and complex NDSRIs | LOD: 0.03\text{ ng/mL} LOQ: 0.1\text{ ng/mL} | 70% – 130% recovery at trace-level spikes | Exceptional MRM transition sensitivity and high-throughput analytical capabilities |
| High-Resolution LC-MS/MS (Orbitrap) | Unknown, non-targeted nitrosamines and structural isomers | LOD: 0.05\text{ ng/mL} LOQ: 0.2\text{ ng/mL} | 70% – 130% recovery at trace-level spikes | Superior mass accuracy that minimizes false positives and supports structural elucidation |
| GC-MS/MS (with Headspace) | Volatile nitrosamines including NDMA, NDEA, and NDIPA | LOD: 0.03\text{ ng/mL} LOQ: $0.1\text{ ng/mL} | 70% – 130% recovery across validated linear range | Reduces injection matrix interference through vapor-phase analysis |
Learn how to engineer your workflows to reliably cross ultra-trace detection thresholds by reading about Ultra-Low Limit of Quantitation (LOQ) in Nitrosamine Testing.
Quantitative Comparison of Permissible Daily Intake Limits
To demonstrate the strict nature of modern nitrosamine regulations, acceptable intake (AI) thresholds established by the FDA, EMA, and Health Canada are calculated based on long-term lifetime cancer risk assessments. The permissible daily intake values for commonly encountered nitrosamine contaminants are structured as follows:
ACCEPTABLE DAILY INTAKE LIMITS (ng/day)
100 ──┬─────────────────────────────────────────────────
│ NDMA: 96 ng/day
│ NMBA: 96 ng/day
80 ──┼─────────────────────────────────────────────────
│
60 ──┼─────────────────────────────────────────────────
│
40 ──┼─────────────────────────────────────────────────
│
20 ──┼ NDEA: 26.5 ng/day
│ DIPNA: Similar strict limits
0 ──┴─────────────────────────────────────────────────When multiple nitrosamines are identified within the same product, the total cumulative intake must remain below 26.5\text{ ng/day} unless compound-specific toxicological data support a higher acceptable limit. For unknown nitrosamine species, analytical methods are generally expected to target the Threshold of Toxicological Concern (TTC), which is established at the extremely low threshold of 18\text{ ng/day}.
Compare global regulatory thresholds and see how limits stack up across jurisdictions at the Nitrosamine AI Limits Comparison Matrix.
Detailed Project Timelines and Milestone Projections
A standard nitrosamine testing program is typically completed within approximately two to six weeks, depending largely on whether the project involves established compendial analytes or highly specialized custom Nitrosamine Drug Substance-Related Impurities. Standard analytical validations are generally completed rapidly, while custom synthesis and structural characterization of uncommon reference standards significantly extend development timelines.
To align analytical activities with product submission schedules and regulatory obligations, including Health Canada’s CAPA implementation deadlines extending through August 1, 2028, project managers must maintain careful oversight of milestone planning and deliverable tracking. A representative timeline projection for a nitrosamine testing project is summarized below.
Project Milestones and Timeframes
| Project Phase / Milestone | Estimated Duration | Core Deliverables & Operational Focus |
| Phase 1: Risk Assessment & Protocol Design | 1 – 3 Business Days | Compilation of synthetic route evaluations, material risk reviews, and formal validation protocols |
| Phase 2a: Standard Method Development | 5 – 10 Business Days | Optimization of LC or GC chromatographic conditions, MS source tuning, and sample preparation workflows |
| Phase 2b: Custom NDSRI Synthesis | 15 – 20 Business Days | Multi-step organic synthesis, purification, NMR validation, and certificate of analysis generation |
| Phase 3: Formal Method Validation | 5 – 10 Business Days | Execution of specificity, linearity, accuracy, precision, LOQ, and robustness validation studies |
| Phase 4: Confirmatory & Stability Testing | 3 – 7 Business Days | Validation batch analysis, sample extraction procedures, and ultra-trace quantitative testing |
| Phase 5: Quality Review & Report Issuance | 3 – 5 Business Days | QA review, audit trail verification, and preparation of submission-ready regulatory documentation |
Exhaustive Cost Structure and Core Financial Expectations
The total cost associated with a comprehensive nitrosamine testing project may range from approximately $1,000 for limited verification studies to more than $35,000 for highly complex, multi-analyte validation programs. Final project expenditures depend on analytical complexity, extraction requirements, instrument runtime, and the need for custom synthesis of specialized reference materials.
When budgeting for outsourced analytical services, pharmaceutical sponsors must carefully evaluate each individual cost component to avoid unforeseen expenses. Full ICH-compliant validations require substantial instrument utilization, laboratory consumables, scientific labor, and administrative oversight. A detailed overview of common pricing categories is provided below.
Comprehensive Cost Breakdown for Nitrosamine Testing Services
| Service Component | Cost Range (USD) | Detailed Technical Breakdown & Resource Allocation |
| Comprehensive Method Validation | $15,000 – $30,000 | Full ICH-compliant validation including statistical reporting, QA review, and regulatory documentation |
| Standard Method Transfer | $12,000 – $22,000 | Bridging studies, matrix compatibility assessments, and equivalency verification at receiving laboratories |
| Partial Method Verification | $1,000 – $6,000 | Limited evaluation of critical parameters for established compendial methods |
| Confirmatory Testing (Per Compound) | $5,000 – $8,000 | Ultra-trace extraction procedures, triple quadrupole analysis, and quantitative reporting |
| NDSRI Custom Synthesis | $15,000 – $20,000+ | Research synthesis, purification, and molecular characterization using NMR, MS, and HPLC |
| Instrument Runtime & Operation | $150 – $300 per hour | Mass spectrometer runtime, mobile phase preparation, column conditioning, and analyst setup |
| Certified Reference Standards | $300 – $2,000 per compound | High-purity analytical reference standards with complete traceability documentation |
| Isotope-Labeled Internal Standards | $200 – $800 per compound | Carbon-13 or deuterated standards used to compensate for matrix-related suppression effects |
| Instrument Qualification (IQ/OQ/PQ) | $7,500 – $15,000 | Installation, operational, and performance qualification supporting cGMP compliance |
| Regulatory Documentation & Reporting | $3,000 – $4,000 | Protocol preparation, statistical review, and final regulatory submission support |
| OOS Out-of-Specification Investigations | $2,000 – $5,000 | Root cause investigations, laboratory deviation analysis, and CAPA implementation |
E-E-A-T Criteria for CRO Selection and Quality Assurance
Assessing a contract laboratory’s experience, expertise, authoritativeness, and trustworthiness (E-E-A-T) requires a detailed review of the organization’s regulatory inspection history, analytical infrastructure, and data integrity practices. Collaborating with a qualified CRO such as ResolveMass Laboratories Inc. provides pharmaceutical developers with access to more than 12 years of advanced mass spectrometry expertise and data management systems aligned with 21 CFR Part 11 requirements.
To ensure the generation of reliable, compliant, and scientifically defensible analytical data, quality assurance teams must perform extensive due diligence during CRO selection. Regulatory agencies routinely inspect laboratory operations, making it critical to partner with organizations that possess established audit histories and proven compliance performance.
CRO DUE DILIGENCE CRITERIA
Experience ==> 12+ years in complex mass spectrometry and impurity analysis
Expertise ==> Validated LC-MS/MS and GC-MS/MS platforms with ppt-level LOQs
Authoritativeness ==> Clean regulatory inspection histories (FDA, Health Canada, EMA)
Trustworthiness ==> Strict adherence to 21 CFR Part 11 and ALCOA+ data guidelines Critical Areas for Evaluating a Potential CRO Include:
Regulatory Inspection Track Record:
Review previous inspection outcomes and determine whether regulatory agencies have raised concerns regarding the laboratory’s nitrosamine data, analytical procedures, or validation methodologies.
Advanced Instrumentation and Sensitivity:
Confirm that the CRO operates modern, qualified analytical platforms such as Triple Quadrupole and high-resolution Orbitrap systems capable of achieving validated signal-to-noise performance at the established Limit of Quantification.
Data Integrity and Security Guidelines:
Ensure that the laboratory enforces secure access controls, immutable audit trails, validated backup systems, and comprehensive adherence to ALCOA+ data integrity principles.
Scientific Partnership and Regulatory Support:
Select a CRO capable of supporting root cause investigations, designing long-term control strategies, and responding effectively to technical inquiries from regulatory authorities.
Concluding Framework for Outsourcing Nitrosamine Testing to a CRO
Successfully controlling the risk associated with mutagenic impurities requires close scientific collaboration with an experienced contract testing organization capable of performing ultra-trace analytical testing within clearly defined operational and regulatory frameworks. Establishing a comprehensive project scope, implementing robust validation protocols, and maintaining transparent pricing structures are all essential for ensuring sustained regulatory compliance and protecting patient safety.
By partnering with an established provider such as ResolveMass Laboratories Inc., pharmaceutical manufacturers gain access to advanced analytical technologies, including LC-MS/MS, GC-MS/MS, and high-resolution Orbitrap mass spectrometry platforms capable of detecting impurities at parts-per-trillion concentrations. Operating under stringent cGMP requirements and comprehensive data integrity standards, ResolveMass delivers scientifically defensible validation packages that satisfy global regulatory expectations.
For pharmaceutical sponsors seeking assistance with defining analytical scopes, planning project timelines, or obtaining detailed pricing proposals for upcoming regulatory submissions, the scientific team at ResolveMass remains available for consultation. To initiate a project discussion or request an analytical proposal, please visit the ResolveMass Laboratories Inc. contact page.
Frequently Asked Questions
The primary reason pharmaceutical companies outsource nitrosamine testing to a CRO is the requirement for highly specialized analytical expertise and ultra-sensitive instrumentation capable of detecting impurities at trace concentrations. Most internal quality control laboratories do not possess advanced systems such as Triple Quadrupole LC-MS/MS or High-Resolution Orbitrap mass spectrometers necessary for parts-per-billion or parts-per-trillion analysis. Contract research organizations provide validated methodologies, experienced scientific teams, and regulatory-compliant workflows aligned with FDA, EMA, and Health Canada requirements. Outsourcing also allows manufacturers to avoid the substantial financial burden associated with purchasing, maintaining, and qualifying sophisticated analytical equipment.
Acceptable intake (AI) limits for newly identified Nitrosamine Drug Substance-Related Impurities (NDSRIs) are established using the Carcinogenic Potency Categorization Approach (CPCA). This scientific framework evaluates the structural characteristics of the impurity, including functional groups, steric hindrance, and molecular features surrounding the N-nitroso moiety, to estimate carcinogenic potency. Based on these structural assessments, compounds are assigned to specific potency categories with corresponding acceptable daily intake thresholds. The resulting AI values may range from $26.5\text{ ng/day}$ for highly potent compounds up to $1,500\text{ ng/day}$ for lower-risk impurities lacking long-term carcinogenicity data.
Validation studies for active pharmaceutical ingredients (APIs) primarily focus on process-related impurities and typically involve simpler analytical matrices with fewer interference challenges. In contrast, finished drug products such as tablets, capsules, and injectable formulations contain complex excipient systems that may create significant matrix effects during analysis. Drug product validations must also assess degradation pathways, extraction recovery, and interactions between packaging materials and the formulation itself. Because of these additional variables, finished product validations generally require more extensive optimization, robustness testing, and matrix-specific recovery evaluations.
High-resolution mass spectrometry (HRMS), including technologies such as Q-Exactive Orbitrap systems, plays a critical role in advanced nitrosamine analysis because it provides exceptional mass accuracy and compound selectivity. Complex pharmaceutical formulations often contain co-eluting compounds with similar nominal masses that may interfere with standard mass spectrometric analysis. HRMS enables precise differentiation between target nitrosamines and unrelated background compounds, significantly reducing the risk of false-positive results. This level of analytical precision is especially important when identifying unknown Nitrosamine Drug Substance-Related Impurities and supporting regulatory submissions.
Nitrosamine contamination associated with pharmaceutical packaging is commonly caused by the migration of secondary amines, residual nitrites, and chemically reactive additives from packaging materials into the drug product. Components such as elastomeric closures, PVC-PVdC blister films, adhesives, and printing lacquers may contain reactive substances capable of participating in nitrosation reactions. Under elevated temperature and high-humidity storage conditions, these chemical precursors can migrate into the formulation through direct contact or microscopic defects in the packaging system. Once present within the product matrix, they may react to form carcinogenic N-nitroso compounds over time.
If analytical testing confirms nitrosamine concentrations above the established acceptable intake limit, pharmaceutical manufacturers are required to immediately notify the relevant regulatory authorities, including the FDA, EMA, or Health Canada. A formal root cause investigation must then be initiated to identify the origin of the contamination and evaluate potential patient risk. Manufacturers are also expected to implement Corrective and Preventive Actions (CAPA) to eliminate or control the source of impurity formation. Depending on the severity of the issue and clinical risk assessment, regulatory consequences may include product recalls, warning letters, or revisions to market authorization status.
Contract research organizations minimize matrix suppression and signal interference by implementing advanced sample cleanup and extraction procedures prior to mass spectrometric analysis. Techniques such as solid-phase extraction (SPE), liquid-liquid extraction, and disposable pipette extraction help remove interfering excipients and matrix components from pharmaceutical samples. Analytical chemists also optimize chromatographic parameters, including gradient profiles, column chemistry, and mobile phase composition, to improve analyte separation. In addition, stable isotope-labeled internal standards are incorporated into the analytical method to compensate for any remaining signal suppression and improve quantification accuracy.
A validated nitrosamine method transfer requires a comprehensive documentation package that demonstrates analytical consistency between the transferring and receiving laboratories. Essential records include approved transfer protocols, system suitability evaluations, comparative precision and accuracy studies, and a finalized transfer report summarizing all findings. Complete analytical raw data, including chromatograms, calibration curves, integration settings, and instrument parameters, must also be retained for regulatory review. All documentation must comply with 21 CFR Part 11 requirements and ALCOA+ data integrity principles to ensure audit readiness and full regulatory traceability.
Reference:
- United States Pharmacopeia. (n.d.). Part one: Industry examining FDA guidance on nitrosamines. USP Quality Matters. https://qualitymatters.usp.org/part-one-industry-examining-fda-guidance-nitrosamines
- Health Canada. (n.d.). Nitrosamine impurities in drugs. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities.html
- Health Canada. (n.d.). Guidance on nitrosamine impurities in medications. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities/medications-guidance.html
- National Center for Biotechnology Information. (2025). Nitrosamine impurities in pharmaceuticals: Analytical challenges and regulatory considerations. PubMed Central. https://pmc.ncbi.nlm.nih.gov/articles/PMC12124699/

