
Introduction:
Peptide therapeutics are one of the fastest-growing drug classes in the pharmaceutical pipeline, but their structural complexity demands rigorous analytical characterization at every stage of development. Two techniques sit at the center of that characterization: peptide mapping and de novo sequencing. While both rely on mass spectrometry and both examine the same fragmented peptide, they answer fundamentally different questions — and choosing the wrong one for a given regulatory or development milestone can cost weeks of rework. This article breaks down when each method applies, how they complement each other, and what a biosimilar or novel peptide developer needs to know before commissioning either service. For a broader overview of how these techniques fit into a full analytical program, see our guide to peptide mapping and sequence analysis.
Summary:
- Peptide mapping confirms that a peptide’s sequence matches its expected reference structure — it’s a verification tool, not a discovery tool.
- De novo sequencing determines an unknown amino acid sequence from scratch, without relying on a reference database or expected sequence.
- Peptide mapping is the standard for lot release, biosimilar comparability, and QC under ICH Q6B; de novo sequencing is used for novel peptide discovery, impurity identification, and unexpected mass shifts.
- Both techniques use enzymatic or chemical digestion followed by LC-MS/MS, but data interpretation workflows differ significantly.
- Many therapeutic peptide programs need both methods at different stages — mapping for routine confirmation, de novo sequencing when mapping reveals unassigned peaks or unexpected fragments.
- Regulatory submissions (FDA, EMA) typically require peptide mapping data as part of the CMC package, with de novo sequencing supporting impurity characterization when needed.
1: What Is Peptide Mapping?
Peptide mapping is an analytical technique that confirms a peptide’s primary structure by comparing its enzymatic or chemical digestion fragments against a known, expected sequence. It answers the question “does this peptide match what it’s supposed to be?” rather than “what is this peptide?”
The process typically involves:
- Enzymatic digestion (commonly trypsin, but also chymotrypsin, Glu-C, or Asp-N depending on the peptide’s sequence and cleavage sites) — see our detailed breakdown of GLP-1 enzymatic digestion mapping
- LC-MS/MS analysis of the resulting fragments
- Comparison against a reference map generated from the known or intended sequence
- Confirmation of post-translational modifications (PTMs), disulfide bonds, and sequence variants
Because the expected sequence is already known, data analysis software can rapidly match observed fragment masses to predicted fragments, flagging any discrepancies as potential impurities, deamidation, oxidation, or truncation products.
2: What Is De Novo Sequencing?
De novo sequencing determines a peptide’s amino acid sequence directly from tandem mass spectrometry fragmentation data, without comparing results to a pre-existing reference sequence. This makes it the method of choice when the sequence is unknown, when a novel peptide is being characterized for the first time, or when an unexpected impurity needs identification.
In de novo sequencing, software (or a trained analyst) interprets the mass differences between consecutive fragment ions (b-ions and y-ions in CID/HCD fragmentation) to reconstruct the amino acid sequence one residue at a time. This is computationally and analytically more demanding than peptide mapping because there is no reference to anchor the interpretation.
3: Peptide Mapping vs De Novo Sequencing: Key Differences
The core distinction between peptide mapping and de novo sequencing comes down to whether a reference sequence exists and what question the analysis is meant to answer. Comparing peptide mapping against related techniques like intact mass analysis can further clarify where each method fits in a characterization workflow.
| Parameter | Peptide Mapping | De Novo Sequencing |
|---|---|---|
| Primary purpose | Confirm known sequence matches reference | Determine unknown sequence from raw data |
| Reference sequence required | Yes | No |
| Typical use case | QC, lot release, biosimilar comparability | Novel peptide discovery, impurity ID |
| Regulatory application | ICH Q6B identity/purity testing | Impurity characterization, root-cause investigation |
| Data interpretation | Automated matching against expected masses | Manual or algorithmic fragment-by-fragment reconstruction |
| Turnaround complexity | Lower — reference-guided | Higher — requires expert interpretation |
| Detects PTMs and variants | Yes, when they deviate from reference | Yes, inherently, since no assumptions are made |
| Common instrumentation | LC-MS/MS (Orbitrap, QTOF) | High-resolution LC-MS/MS with advanced fragmentation |
4: When Should You Use Peptide Mapping?
Peptide mapping is the right choice whenever you already know — or believe you know — what the peptide sequence should be and need to confirm it. This is why it’s a standard component of routine quality control and regulatory submissions, and why it plays such a central role in peptide mapping for biosimilars.
Common applications include:
- Batch-to-batch consistency testing during manufacturing
- Biosimilar comparability studies, confirming the biosimilar’s sequence matches the reference product
- Identity and purity testing required under ICH Q6B guidelines for peptide and protein drug substances, including GLP-1 peptide mapping regulatory requirements
- PTM monitoring, such as tracking oxidation, deamidation, or disulfide bond formation across production lots
- Stability studies, where mapping is repeated over time to detect degradation-related sequence changes
Because the reference sequence is already established, peptide mapping delivers faster, more reproducible results suited to the repetitive testing cadence of GMP manufacturing environments. This is especially true for GLP-1 class peptides, where GLP-1 peptide mapping and characterization has become a routine part of generic and biosimilar development programs.
5: When Should You Use De Novo Sequencing?
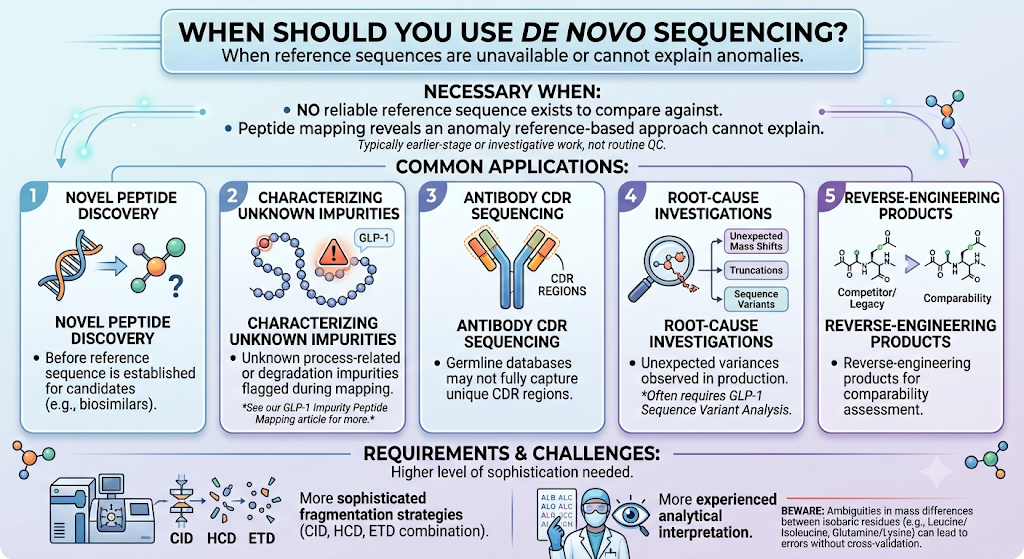
De novo sequencing becomes necessary when there is no reliable reference sequence to compare against, or when peptide mapping reveals an anomaly that the reference-based approach cannot explain. This typically arises in earlier-stage or investigative work rather than routine QC.
Common applications include:
- Novel peptide or biosimilar candidate discovery, before a reference sequence has been fully established
- Characterizing unknown process-related or degradation impurities flagged during mapping but not matching any predicted fragment — a workflow covered in depth in our article on GLP-1 impurity peptide mapping
- Antibody and antibody-fragment sequencing where germline databases may not fully capture unique CDR regions
- Root-cause investigations into unexpected mass shifts, truncations, or sequence variants observed in production, often requiring GLP-1 sequence variant analysis
- Reverse-engineering competitor or legacy peptide products for comparability assessment
De novo sequencing requires more sophisticated fragmentation strategies (often combining CID, HCD, and ETD) and more experienced analytical interpretation, since even small ambiguities in mass differences between isobaric or near-isobaric residues (such as leucine/isoleucine or glutamine/lysine) can lead to sequencing errors without careful cross-validation.
6: Do Therapeutic Peptide Programs Need Both Methods?
Yes — most mature therapeutic peptide programs use peptide mapping for routine confirmation and reserve de novo sequencing for situations mapping cannot resolve. The two methods are complementary rather than interchangeable.
A typical workflow looks like this:
- Early discovery/development — de novo sequencing establishes or confirms the peptide’s structure when no validated reference exists yet.
- Reference sequence lock — once the sequence is confirmed, it becomes the baseline for all future mapping, as demonstrated in our semaglutide peptide mapping work.
- Routine QC and manufacturing — peptide mapping verifies each batch against that locked reference, following the same principles outlined across our GLP-1 peptide mapping case studies.
- Anomaly investigation — if mapping detects an unassigned peak or unexpected mass, de novo sequencing is used to characterize the impurity.
This layered approach ensures both regulatory compliance and scientific rigor: mapping keeps routine testing efficient, while de novo sequencing provides the depth needed when something falls outside expected parameters.
7: How Does This Apply to Biosimilar Peptide Development?
For biosimilar peptide development, peptide mapping vs de novo sequencing decisions are shaped by regulatory expectations from both the FDA and EMA, which require demonstrated sequence identity to the reference product as part of the analytical similarity package. Peptide mapping is the primary tool for this comparability demonstration, since the reference product’s sequence is already published or well-characterized.
This is especially relevant across the GLP-1 and generic peptide space, where ResolveMass has supported sameness and comparability studies for a wide range of therapeutic peptides, including semaglutide, tirzepatide, liraglutide, dulaglutide, exenatide, retatrutide, and lanreotide. Similar principles apply to other complex peptide-based drug products, such as insulin analog characterization for generic ANDA submissions.
De novo sequencing plays a supporting role — for instance, when a biosimilar developer needs to independently verify a reference product’s sequence from purchased reference standard material, or when unexpected PTM patterns emerge during head-to-head comparison that require deeper structural investigation beyond simple mass matching.
8: Choosing the Right Analytical Partner
Selecting between peptide mapping and de novo sequencing — or determining when a program needs both — depends on the specific development stage, regulatory pathway, and the complexity of the peptide’s structure, including any non-standard amino acids, cyclic structures, or complex PTM profiles. Working with an analytical partner experienced across both reference-based and de novo workflows helps avoid delays caused by choosing an insufficient method for a given regulatory milestone.
ResolveMass Laboratories supports therapeutic peptide developers with both peptide mapping and de novo sequencing services, backed by high-resolution LC-MS/MS platforms and analytical teams experienced in ICH Q6B-compliant identity testing, biosimilar comparability studies, and impurity characterization. Whether your program requires routine lot release testing or in-depth structural investigation of an unknown impurity, aligning the right method to the right development stage is critical to staying on schedule and audit-ready.
Conclusion:
The distinction between peptide mapping and de novo sequencing comes down to one question: do you already know what sequence you’re looking for? Peptide mapping confirms a known structure against a reference and anchors routine QC and biosimilar comparability testing, while de novo sequencing reconstructs an unknown sequence from raw fragmentation data and supports discovery-stage work and impurity investigations. Most therapeutic peptide programs rely on both at different points in the development lifecycle — mapping for consistency, de novo sequencing for discovery and troubleshooting. Understanding when each applies helps development teams plan analytical timelines accurately and avoid costly method-selection missteps.
Frequently Asked Questions:
Peptide mapping is highly accurate for confirming the identity of therapeutic peptides when the amino acid sequence is already known. It compares experimentally generated peptide fragments with a theoretical reference sequence using high-resolution LC-MS/MS. The technique can achieve excellent sequence coverage while detecting amino acid substitutions, truncations, and post-translational modifications. It also helps verify batch-to-batch consistency and product integrity throughout development. Because of its reliability, peptide mapping is widely used in quality control and regulatory submissions. When performed using validated methods, it provides strong confidence in peptide identity and structural characterization.
De novo sequencing can analyze a wide variety of peptide samples, including purified therapeutic peptides, biological extracts, degradation products, synthetic peptides, and unknown impurities. It is especially valuable when no reference sequence is available or when database matching is not possible. The technique reconstructs the amino acid sequence directly from tandem mass spectrometry data. It can also be applied to natural peptide discovery and reverse engineering studies. High-quality sample preparation and fragmentation data are essential for obtaining accurate sequence information. This flexibility makes de novo sequencing an important tool in pharmaceutical research.
Enzymatic digestion is commonly used in peptide mapping, particularly for large proteins or long peptide chains, to generate smaller fragments suitable for LC-MS/MS analysis. Enzymes such as trypsin, Lys-C, or Glu-C are selected based on the analytical objective. However, shorter therapeutic peptides may sometimes be analyzed without digestion if they are compatible with direct mass spectrometric analysis. The choice depends on peptide size, complexity, and required sequence coverage. Proper digestion improves fragment identification and enhances analytical confidence. Optimized digestion protocols are therefore an important part of peptide mapping workflows.
Although de novo sequencing is highly powerful, it does have certain limitations. The accuracy of sequence reconstruction depends heavily on the quality of MS/MS fragmentation spectra and instrument resolution. Incomplete fragmentation or low-abundance ions can make sequence interpretation more difficult. Some amino acids, such as leucine and isoleucine, have identical masses and cannot always be distinguished by mass spectrometry alone. Complex post-translational modifications may also complicate data analysis. Therefore, complementary analytical techniques are often used to confirm the final sequence assignment.
Yes. De novo sequencing is capable of identifying sequence variants that may not be detected through conventional database searches. It reconstructs the peptide sequence directly from fragmentation data, allowing scientists to identify amino acid substitutions, insertions, deletions, and unexpected sequence changes. This capability is especially useful when investigating impurities, degradation products, or manufacturing-related variants. The technique can also support reverse engineering of competitor products. High-resolution mass spectrometry and advanced software improve confidence in sequence assignments. Additional analytical confirmation may be performed when required.
The duration of a peptide mapping study depends on sample complexity, project scope, and the level of characterization required. Routine identity confirmation may be completed within a few days, while comprehensive studies involving multiple batches, stability testing, or post-translational modification analysis can take several weeks. Sample preparation, LC-MS/MS analysis, data interpretation, and quality review all contribute to the overall timeline. Method development may extend the project duration for challenging samples. Proper planning ensures efficient execution while maintaining data quality and regulatory compliance.
De novo peptide sequencing relies on specialized bioinformatics software that interprets tandem mass spectrometry fragmentation data to reconstruct amino acid sequences. These programs analyze fragmentation patterns, generate sequence candidates, and assign confidence scores for each result. The software also assists in identifying sequence variants and unknown modifications. However, automated results are typically reviewed by experienced mass spectrometry scientists to ensure accuracy. Manual verification is particularly important for complex peptides and low-quality spectra. Combining advanced algorithms with expert interpretation produces the most reliable sequence assignments.
Yes. Peptide mapping is widely used to monitor structural changes that occur during forced degradation and long-term stability studies. It can detect oxidation, deamidation, hydrolysis, disulfide bond rearrangement, and peptide truncation, all of which may affect therapeutic performance. By comparing stressed samples with the original reference material, scientists can identify degradation pathways and evaluate product stability. This information supports formulation optimization, shelf-life determination, and regulatory documentation. Peptide mapping therefore plays a key role in stability assessment throughout the product lifecycle.
Reference
- Muth T, Hartkopf F, Vaudel M, Renard BY. A potential golden age to come—current tools, recent use cases, and future avenues for de novo sequencing in proteomics. Proteomics. 2018 Sep;18(18):1700150.https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/abs/10.1002/pmic.201700150
- O’Bryon I, Jenson SC, Merkley ED. Flying blind, or just flying under the radar? The underappreciated power of de novo methods of mass spectrometric peptide identification. Protein Science. 2020 Sep;29(9):1864-78.https://onlinelibrary.wiley.com/doi/abs/10.1002/pro.3919
- Cifani P, Dhabaria A, Chen Z, Yoshimi A, Kawaler E, Abdel-Wahab O, Poirier JT, Kentsis A. ProteomeGenerator: a framework for comprehensive proteomics based on de novo transcriptome assembly and high-accuracy peptide mass spectral matching. Journal of proteome research. 2018 Oct 8;17(11):3681-92.https://pubs.acs.org/doi/abs/10.1021/acs.jproteome.8b00295
- Bandeira N, Pham V, Pevzner P, Arnott D, Lill JR. Automated de novo protein sequencing of monoclonal antibodies. Nature biotechnology. 2008 Dec;26(12):1336-8.https://www.nature.com/articles/nbt1208-1336
- Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SV, Kaas Q, Eletsky A, Huang PS. Accurate de novo design of hyperstable constrained peptides. Nature. 2016 Oct 20;538(7625):329-35.https://www.nature.com/articles/nature19791

