
Introduction
Peptide physicochemical characterization services establish the scientific basis required to confirm the molecular identity, therapeutic suitability, and regulatory compliance of peptide-based drug candidates. These comprehensive characterization strategies evaluate the thermodynamic, physical, and chemical properties of peptide molecules, supporting their successful progression from early-stage research through commercial-scale manufacturing. According to the regulatory guidelines established by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), biopolymers containing 40 or fewer amino acids are classified as peptides. Based on this regulatory definition, synthetic peptides may qualify for the Abbreviated New Drug Application (ANDA) pathway under Section 505(j) of the FD&C Act, provided they demonstrate structural and chemical “sameness” to a recombinant reference listed drug (RLD). Because peptides possess inherent structural complexity and are highly susceptible to both physical and chemical degradation, comprehensive analytical validation performed by specialized contract research organizations (CROs) is essential. The combined application of orthogonal analytical technologies, including high-resolution mass spectrometry (LC-HRMS), capillary zone electrophoresis (CZE), and nuclear magnetic resonance (NMR) spectroscopy, enables the accurate evaluation of critical quality attributes (CQAs) that directly influence clinical safety, pharmacokinetic behavior, and overall product shelf-life.
Explore our specialized Peptide Sameness Study Services in the United States to support your ANDA submissions.
Share via:
Article Summary:
- Peptide physicochemical characterization provides essential analytical data to verify peptide identity, quality, stability, and regulatory compliance, supporting drug development from early research through commercialization.
- Solubility profiling evaluates how peptides behave under different pH and solvent conditions, helping scientists select optimal formulation strategies that minimize aggregation and maximize solution stability.
- pKa determination uses advanced analytical techniques to measure peptide ionization properties, enabling accurate prediction of molecular charge, solubility, receptor interactions, and formulation performance across varying pH levels.
- Log P and Log D analysis assesses peptide lipophilicity to estimate membrane permeability, tissue distribution, and bioavailability, providing critical insights for drug absorption and pharmacokinetic optimization.
- Stability profiling identifies both chemical degradation (such as oxidation, deamidation, and isomerization) and physical changes (including unfolding and aggregation), ensuring long-term product quality and shelf-life.
- Multiple complementary analytical technologies, including chromatography, mass spectrometry, electrophoresis, spectroscopy, and biophysical methods, work together to generate reliable, scientifically robust characterization data.
- Comprehensive physicochemical characterization plays a vital role in meeting FDA, EMA, and ICH regulatory expectations by demonstrating peptide quality, generic sameness, and manufacturing consistency while reducing development risks and accelerating regulatory approval.
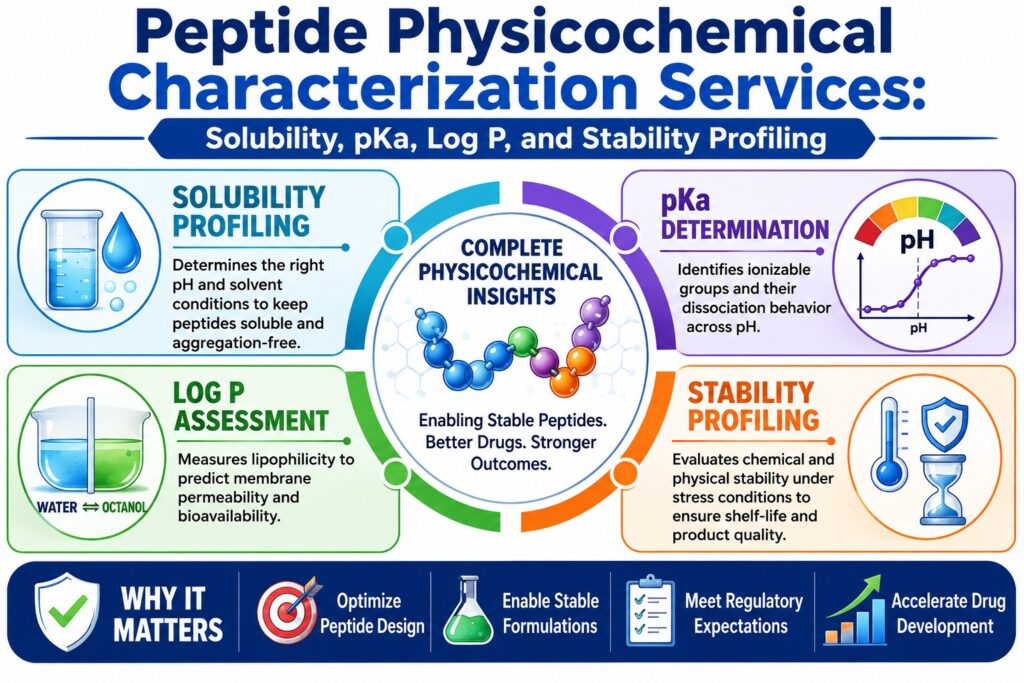
Why are Peptide Physicochemical Characterization Services Essential in Drug Development?
Peptide physicochemical characterization services play a fundamental role in drug development by minimizing the clinical and manufacturing risks associated with peptide micro-heterogeneity, aggregation, and chemical instability. Through the generation of quantitative biophysical data, these services help pharmaceutical developers formulate stable products, establish critical quality attributes, and fulfill the regulatory expectations required for global product approvals.
Peptides represent a distinctive class of therapeutic molecules that bridge the gap between conventional small molecules and large biologic proteins. They combine the exceptional target specificity typically associated with biologics with the well-defined chemical synthesis and manufacturing processes characteristic of small-molecule drugs. Nevertheless, their intricate primary structures and conformational flexibility create considerable analytical challenges throughout development. Unlike traditional small molecules, peptides are highly responsive to environmental conditions such as pH, ionic strength, temperature, and mechanical shear stress. Variations in these factors can rapidly induce physical aggregation or initiate chemical degradation, potentially affecting both product quality and therapeutic performance.
Implementing a comprehensive physicochemical characterization strategy during the early stages of development allows researchers to identify potential formulation liabilities, optimize lead peptide sequences, and generate scientifically robust datasets suitable for regulatory submissions, including Investigational New Drug (IND) applications, New Drug Applications (NDA), and generic Abbreviated New Drug Applications (ANDA). These analytical services are especially important for structurally complex and extensively modified peptide therapeutics, including cyclic peptides, peptide-drug conjugates (PDCs), PEGylated peptides, and lipidated glucagon-like peptide-1 (GLP-1) receptor agonists such as semaglutide, liraglutide, and tirzepatide.
Learn more about our LC-MS Characterization of GLP-1 Peptides and ensure the precision of your therapeutic candidates.
How is Solubility Profiling Conducted within Peptide Physicochemical Characterization Services?
Within peptide physicochemical characterization services, solubility profiling is performed by determining a peptide’s net electrical charge across different pH conditions and applying a structured solvent selection strategy. This systematic experimental workflow identifies the precise concentration limits and pH-solvent combinations required to maintain the peptide as a stable, unaggregated monomer in solution.
For most therapeutic applications, peptide formulations are typically developed to achieve concentrations between 1 and 2 mg/mL. Maintaining this concentration range helps prevent molecular crowding while ensuring analytical accuracy during both in vitro and in vivo investigations. Peptide solubility is primarily influenced by amino acid composition, sequence length, and the balance between hydrophilic and hydrophobic side chains. Before laboratory reconstitution, the theoretical net charge of the peptide is estimated by assigning charge values to ionizable residues under physiological pH conditions:
- Acidic Residues: Aspartic acid (D), glutamic acid (E), and the C-terminal carboxyl group (COOH) are assigned a charge value of -1.
- Basic Residues: Lysine (K), arginine (R), and the N-terminal amino group (NH₂) are assigned a charge value of +1.
- Histidine: Assigned a charge value of +1 at pH < 6 and 0 at pH > 6.
The total of these individual charges determines the peptide’s isoelectric point (pI), which represents the pH at which the molecule carries no overall electrical charge. At this point, electrostatic repulsion between peptide molecules is significantly reduced, making the peptide highly susceptible to rapid aggregation and gel formation when the surrounding solvent pH approaches its calculated pI. Consequently, standard formulation practices recommend maintaining buffer pH values at least one pH unit above or below the peptide’s calculated pI to preserve solution stability and minimize aggregation.
For peptides containing a high proportion of hydrophobic amino acids (≥ 50% hydrophobic residues, including Val, Leu, Ile, Phe, Trp, Met, and Tyr), conventional aqueous buffers generally do not provide adequate solubility. In such cases, organic co-solvents such as dimethyl sulfoxide (DMSO), dimethylformamide (DMF), or acetonitrile (ACN) are incorporated to improve peptide dissolution. Similarly, peptides with structures predominantly composed of β-sheets often require the use of chaotropic denaturing agents, including 6 M guanidine hydrochloride or 8 M urea, to temporarily disrupt intermolecular hydrogen bonding and promote efficient solubilization.
Peptide Classification and Recommended Reconstitution Strategies
| Peptide Classification | Net Charge Criteria (at pH 7.0) | Theoretical Charge Ratio | Primary Reconstitution Strategy | Secondary Co-solvent / Modifier |
|---|---|---|---|---|
| Acidic Peptide | Net negative charge | > 25% of residues are D and E | Reconstitute in a minimal volume of 0.1 M Ammonium Bicarbonate or 0.1% Ammonium Hydroxide | Dilute gradually with sterile water or phosphate-buffered saline (PBS) until the desired final volume is achieved. |
| Basic Peptide | Net positive charge | 10–25% of residues are K and R | Reconstitute in a minimal volume of 0.1% to 25% Acetic Acid | Dilute with physiological PBS maintained at pH 7.0–7.4. |
| Neutral / Polar Peptide | Net charge of zero | > 25% charged residues | Reconstitute using pH-adjusted aqueous buffer systems. | Sonicate in a water bath maintained below 40°C to facilitate the dissolution of larger particles. |
| Hydrophobic Peptide | Variable net charge | < 10% charged residues | Dissolve in a minimal volume of 50% (v/v) DMSO, DMF, or ACN. | Slowly dilute into sterile water or an appropriate aqueous buffer while maintaining continuous agitation. |
| Insoluble / Gel-Forming Peptide | Zero net charge or extensive crosslinking | ≥ 75% of the sequence consists of D, E, H, K, N, Q, R, S, T, and Y | Introduce chaotropic agents such as 6 M Guanidine-HCl or 8 M Urea. | If the peptide remains incompatible with downstream bioassays, optimize the sequence by shortening its length or incorporating charged lysine tags. |
For detailed insights into sequence fidelity, visit our Peptide Sequencing of GLP-1 Drugs page.
What Methods Determine Dissociation Constants (pKa) via Peptide Physicochemical Characterization Services?
Within peptide physicochemical characterization services, dissociation constants (pKa) are determined using capillary zone electrophoresis (CZE), potentiometric titration, and nuclear magnetic resonance (NMR) spectroscopy to establish the precise ionization characteristics of peptide side chains. These analytical measurements define the overall molecular charge of a peptide across different pH conditions, directly influencing its solubility, membrane permeability, and receptor-binding behavior.
Accurate determination of pKa values in peptides is particularly challenging because peptides are polyprotic systems containing multiple ionizable functional groups with closely overlapping dissociation constants. To overcome these analytical complexities, several complementary thermodynamic and separation-based techniques are employed.
Capillary Zone Electrophoresis (CZE):
Capillary Zone Electrophoresis is widely recognized as one of the most effective techniques for peptide pKa characterization because it measures the effective electrophoretic mobility (μeff) of the peptide as a direct function of pH. During analysis, the peptide sample is introduced into a bare fused silica capillary and exposed to a multi-point buffer gradient, typically consisting of 24 evenly distributed pH values ranging from 1.8 to 11.2. Since the separation mechanism is based entirely on molecular charge, impurities and degradation products do not interfere with the migration behavior of the target analyte. This enables highly accurate pKa determination while requiring only very small sample volumes, approximately 30 μL. The resulting electrophoretic mobility data are subsequently fitted to ionization models to identify the inflection point corresponding to the peptide’s pKa value.
NMR Titration Spectroscopy:
Nuclear magnetic resonance (NMR) titration is considered the gold-standard technique for determining site-specific microscopic pKa values in structurally complex and conformationally folded peptides. This method monitors pH-dependent chemical shifts (δobs) of proton (¹H), carbon (¹³C), or nitrogen (¹⁵N) nuclei located near ionizable functional groups. By observing these chemical shift changes, the protonation state of individual amino acid residues can be accurately determined. Advanced two-dimensional Chemical Shift Imaging (CSI), which establishes a carefully controlled pH gradient throughout a single NMR tube, enables automated “single-shot” pKa determination while preserving sample integrity and significantly reducing analysis time.
Potentiometric Titration:
Potentiometric titration is a classical analytical technique that determines pKa values by measuring changes in the electrical potential (Ecell) generated between a glass electrode and a reference electrode during the controlled addition of standardized acid or base solutions. Although this method provides excellent accuracy for relatively simple and highly soluble peptides, it requires large quantities of highly purified material, typically exceeding 10⁻⁴ M concentration. Furthermore, potentiometric titration demonstrates limited resolution when analyzing multiprotic peptides containing overlapping ionization events or peptides exhibiting poor aqueous solubility.
Comparison of pKa Determination Methods
| Characterization Parameter | Potentiometric Titration | Capillary Zone Electrophoresis (CZE) | NMR Titration (CSI) |
|---|---|---|---|
| Primary Output | Thermodynamic macroscopic pKa | Effective electrophoretic mobility (μeff) versus pH inflection profile | Microscopic, site-specific pKa values of individual residues |
| Sample Volume / Concentration | High (> 10⁻⁴ M; requires large sample volumes) | Ultra-low (approximately 30 μL sample requirement) | Low to moderate sample requirements compatible with micro-probe systems |
| Purity Requirement | Extremely high (> 95% purity required) | Low, because impurities are separated during analysis | Moderate, with tolerance for minor impurities and degradation products |
| pH Operating Range | Typically 2.0 to 12.0 | Broad range of 1.8 to 11.2 using 24-point buffer systems | Broad range of 1.0 to 13.0 using indicator molecules |
| Primary Limitation | Insoluble peptides may cause electrode drift and precipitation | Joule heating at elevated ionic strengths can distort electrophoretic peak shapes | Large and structurally complex peptides may exhibit significant spectral overlap |
Discover our comprehensive CRO for GLP-1 Peptide Characterization and streamline your development pipeline.
How are Log P and Log D Partition Coefficients Measured to Evaluate Peptide Lipophilicity?
Peptide lipophilicity is evaluated using established thermodynamic partitioning techniques, including the shake-flask method (OECD 107), the slow-stirring method (OECD 123), and reversed-phase HPLC retention profiling (OECD 117), to determine Log P and Log D values. These partition coefficients measure the relative affinity of a peptide for lipid environments compared with aqueous phases, making them essential indicators of oral absorption potential, tissue distribution characteristics, and systemic clearance.
Lipophilicity is commonly expressed as the octanol/water partition coefficient (log P) for non-ionizable compounds and as the distribution coefficient (log D) for ionizable molecules measured at a specified pH, most commonly log D₇.₄ under physiological conditions. The determination of these parameters follows internationally recognized analytical guidelines to ensure consistency, reproducibility, and regulatory compliance.
OECD Standard 107 (Shake-Flask Method)
The OECD 107 Shake-Flask Method is regarded as the reference procedure for directly determining peptide partition coefficients. In this method, the peptide is dissolved in a pre-equilibrated biphasic system composed of n-octanol and an aqueous buffer. The mixture is vigorously shaken until thermodynamic equilibrium is established, after which the organic and aqueous phases are separated by centrifugation. The concentration of peptide present in each phase is then quantified using high-performance liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). The distribution coefficient is calculated according to the following equation:
Log D Equation:
log DpH = log ( [peptide]octanol / [peptide]aqueous )
This direct analytical technique provides highly accurate results for peptides with log D values ranging from -2.0 to 4.5. However, the procedure is relatively labor-intensive and becomes unsuitable when peptides possess surface-active characteristics that stabilize emulsions between the two phases.
OECD Standard 123 (Slow-Stirring Method)
The OECD 123 Slow-Stirring Method is specifically designed for highly hydrophobic or lipidated peptides, including semaglutide, liraglutide, and tirzepatide, where log P values exceed 4.5. Under these conditions, the conventional shake-flask technique becomes unreliable because vigorous mixing promotes the formation of persistent micro-emulsions. Instead, the slow-stirring method gently mixes the aqueous and organic phases using minimal agitation, preventing emulsion formation while allowing true thermodynamic equilibrium to develop. Although this approach provides more reliable partition measurements for highly lipophilic peptides, equilibrium generally requires two to three days to reach completion.
OECD Standard 117 (RP-HPLC Chromatographic Method)
The OECD 117 Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC) method offers an indirect yet highly efficient approach for determining peptide lipophilicity. This technique correlates the chromatographic retention time (tR) of a peptide on a hydrophobic stationary phase, typically a C18 column, with its partitioning behavior. Both gradient and isocratic chromatographic conditions may be employed, and retention data are compared with structurally related reference compounds whose partition coefficients have already been established using the shake-flask method. This comparison generates a reliable lipophilicity index. The RP-HPLC approach is particularly advantageous for high-throughput screening because it requires only small sample quantities, supports automated analysis, and remains largely unaffected by sample impurities.
Comparison of Lipophilicity Assays
| Lipophilicity Assay | Standard Regulation | Applicable Log P/D Range | Primary Analytical Strengths | Operational Drawbacks |
|---|---|---|---|---|
| Shake-Flask Method | OECD 107 | -2.0 to 4.5 | Direct thermodynamic measurement and accepted regulatory reference standard | High solvent consumption and emulsion formation with surface-active peptides |
| Slow-Stirring Method | OECD 123 | > 4.5 (highly hydrophobic peptides) | Prevents emulsion formation and is well suited for lipidated peptides | Requires prolonged equilibration times of approximately 2 to 3 days |
| Chromatographic Retention (RP-HPLC) | OECD 117 | Broad operational range | High-throughput analysis, automation compatible, and requires minimal sample volume | Indirect measurement requiring calibration against structurally similar reference compounds |
Address stability concerns early with our GLP-1 Peptide Impurity Characterization services.
What Analytical Techniques Underpin Stability Profiling within Peptide Physicochemical Characterization Services?
Stability profiling within peptide physicochemical characterization services relies on an integrated combination of high-resolution chromatographic separation techniques and advanced mass spectrometric analysis to detect, identify, and monitor both chemical and physical degradation pathways. These complementary analytical platforms provide the structural resolution necessary to establish stability-indicating methods capable of confirming that drug potency, molecular integrity, and overall mass balance remain consistent throughout the product’s intended shelf-life.
Unlike conventional small-molecule therapeutics, peptides are susceptible to numerous degradation mechanisms because they contain multiple reactive side chains, exhibit considerable conformational flexibility, and readily respond to environmental influences. Demonstrating that an analytical method is stability-indicating requires clear evidence that the analytical system can effectively separate the active pharmaceutical ingredient (API) from degradation products, manufacturing-related impurities, and formulation excipients under all anticipated storage conditions.
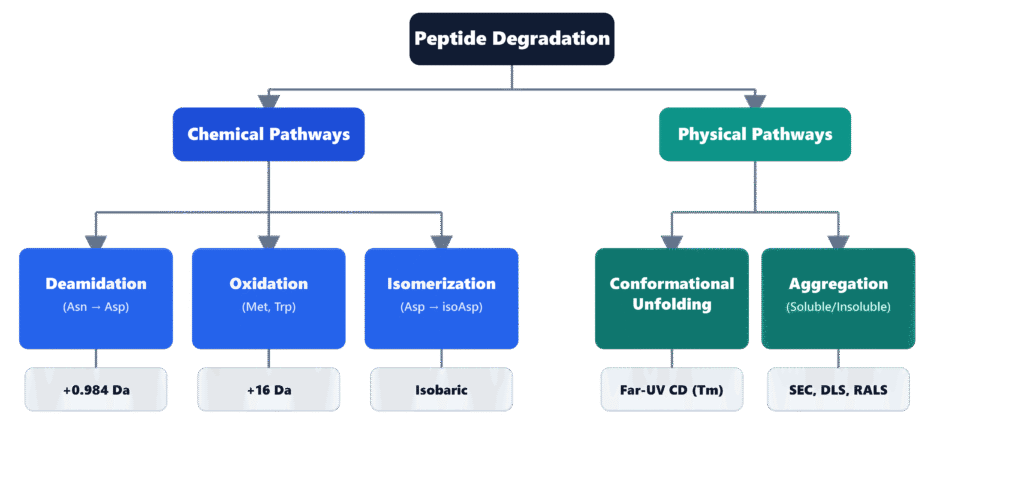
Major Peptide Degradation Pathways
Optimize your storage conditions with our GLP-1 Peptide Stability Analytical Methods.
How is Chemical Degradation Profiled in Peptide Stability Studies?
Chemical degradation in peptide stability studies is characterized through highly selective chromatographic separation and mass-based identification of degradation products resulting from deamidation, oxidation, isomerization, and hydrolysis. By subjecting peptide samples to carefully controlled forced-degradation conditions, scientists can determine the precise kinetics of covalent modifications that may compromise therapeutic efficacy, product quality, or patient safety.
Therapeutic peptides frequently undergo spontaneous non-enzymatic chemical modifications during purification, manufacturing, handling, and long-term storage. Consequently, stability-indicating analytical methods are designed to monitor the principal degradation pathways described below.
Deamidation
Deamidation primarily affects Asparagine (Asn) and Glutamine (Gln) residues. Under neutral or alkaline conditions, the nitrogen atom of the neighboring C-flanking peptide bond attacks the side-chain carbonyl group of Asn, producing a cyclic succinimidyl (Asu) intermediate. Hydrolysis of this intermediate subsequently generates a mixture of normal α-aspartyl and β-aspartyl (isoaspartyl) residues, typically in an approximate ratio of 1:3. This chemical transformation introduces an additional negative charge and produces a monoisotopic mass increase of +0.984 Da, which can be accurately detected using high-resolution Orbitrap mass spectrometry.
Oxidation
Methionine (Met) and Tryptophan (Trp) residues are particularly vulnerable to oxidation caused by dissolved oxygen and reactive peroxide species. Oxidation of Methionine results in the formation of methionine sulfoxide, producing a characteristic mass increase of +15.995 Da. High-resolution mass spectrometry (LC-HRMS), combined with tandem MS/MS fragmentation through b-ion and y-ion analysis, enables precise localization of the oxidation site. These data assist formulation scientists in selecting suitable antioxidant systems and establishing oxygen-controlled storage conditions that minimize oxidative degradation.
Isomerization
Aspartic acid (Asp) residues are capable of spontaneous conversion to isoaspartic acid (isoAsp) through isomerization. Because this structural rearrangement is isobaric, conventional mass spectrometry alone cannot distinguish the modified residue from its native counterpart. Accurate identification therefore requires advanced chromatographic separation techniques, including reversed-phase ultra-high-performance liquid chromatography (RP-UHPLC) using specialized C18 stationary phases or Strong Cation Exchange (SCX) two-dimensional LC-MS. These analytical approaches exploit subtle differences in chromatographic retention behavior and molecular conformation to achieve reliable separation and identification of isoAsp-containing peptide variants.
Stay compliant with our Regulatory Requirements for GLP-1 Peptide Characterization.
What Biophysical Methods Monitor Physical Stability and Aggregation Kinetics?
Physical stability and aggregation kinetics are evaluated using circular dichroism (CD) spectroscopy, dynamic light scattering (DLS), and size-exclusion chromatography (SEC-HPLC) to monitor conformational alterations and molecular self-association. These complementary, non-destructive biophysical techniques are capable of detecting both reversible oligomer formation and irreversible fibrillar aggregation under thermal, mechanical, and environmental stress conditions.
The physical degradation of peptides generally begins with conformational unfolding or disruption of higher-order structures (HOS). As these structural changes occur, hydrophobic regions that were previously buried within the peptide become exposed, promoting intermolecular interactions that lead to the formation of soluble oligomers and, eventually, insoluble fibrillar aggregates. Accurate monitoring of these processes requires the coordinated use of multiple biophysical analytical techniques.
Circular Dichroism (CD) Spectroscopy
Far-UV Circular Dichroism (CD) spectroscopy is employed to monitor changes in peptide secondary structure, including transitions from α-helix to β-sheet conformations, during controlled temperature ramp experiments. This analytical technique provides direct measurement of the thermal denaturation or melting temperature (Tm), which represents the temperature at which 50% of the peptide population has unfolded. Monitoring Tm provides valuable insight into the thermal stability of peptide therapeutics and supports formulation optimization.
Dynamic Light Scattering (DLS)
Dynamic Light Scattering (DLS) measures the hydrodynamic radius (RH) and diffusion coefficient of peptide molecules suspended in solution. These measurements enable researchers to monitor the progression of molecular assembly from individual monomeric peptides to progressively larger oligomeric and macromolecular aggregates. Temperature-dependent DLS experiments are also used to determine the aggregation onset temperature (Tons), an important indicator that correlates closely with the expected physical shelf-life of the peptide formulation.
Size-Exclusion Chromatography (SEC-HPLC)
Size-Exclusion Chromatography (SEC-HPLC) serves as the primary quantitative analytical method for determining the relative proportions of peptide monomers, soluble oligomers, and high-molecular-weight (HMW) species present in stability samples. However, because very large aggregates may either be retained by the pre-column or dissociate during sample dilution before chromatographic injection, SEC results are routinely complemented with Right Angle Light Scattering (RALS) or Nanoparticle Tracking Analysis (NTA). The combined use of these techniques provides a more comprehensive and unbiased assessment of peptide aggregation behavior.
Utilize 2D NMR for Peptide Characterization to gain deeper insights into your molecule’s structure.
How Do Peptide Physicochemical Characterization Services Facilitate ANDA Sameness and FDA/EMA Compliance?
Peptide physicochemical characterization services support ANDA sameness evaluations and regulatory compliance by generating a comprehensive comparative profile between a generic synthetic peptide and its recombinant reference listed drug (RLD). Through orthogonal verification of the primary amino acid sequence, confirmation of higher-order structural similarity, and detailed impurity characterization, these analytical services produce scientifically robust data packages required for successful regulatory submissions.
When sponsors submit an Abbreviated New Drug Application (ANDA) for a generic synthetic peptide referencing a recombinant DNA (rDNA)-derived RLD, including products such as glucagon, liraglutide, nesiritide, teriparatide, or teduglutide, they must demonstrate that the active pharmaceutical ingredient is structurally and chemically identical to the reference product. The FDA’s draft guidance regarding ANDA sameness requires comprehensive analytical evaluation across multiple structural and physicochemical attributes.
Primary Sequence and Physicochemical Properties
Confirmation of identical primary amino acid sequences is achieved through high-resolution peptide mapping. The peptide undergoes enzymatic or chemical cleavage, followed by separation using reversed-phase HPLC and analysis with high-resolution tandem mass spectrometry (LC-HRMS/MS). Evaluation of b-ion and y-ion fragmentation patterns confirms complete sequence fidelity while excluding amino acid substitutions or sequence variations.
Higher-Order Structure (HOS)
Comparative higher-order structure analysis verifies that the synthetic peptide adopts secondary and tertiary conformations equivalent to those of the recombinant reference listed drug. This structural equivalence is established using Far-UV Circular Dichroism (CD) spectroscopy together with high-field one-dimensional and two-dimensional NMR spectroscopy.
Oligomerization and Aggregation States
The aggregation characteristics of the generic peptide must remain comparable to those of the reference product throughout its intended shelf-life. This assessment is performed using SEC-MALS (Size-Exclusion Chromatography coupled with Multi-Angle Light Scattering), analytical ultracentrifugation (AUC), and Thioflavin-T (ThT) fluorescence assays to characterize oligomer formation and aggregate populations.
Salt Form and Counter-Ion Equivalence
Generic peptide products must contain the same salt or ester form as the reference listed drug in equivalent proportions. Residual counter-ions must also be accurately quantified and carefully controlled. Particular attention is given to the selection of counter-ions, such as acetate and trifluoroacetic acid (TFA), because the EMA peptide guideline identifies residual TFA as a potential source of toxicity and stability concerns, requiring appropriate justification and well-defined control strategies.
Regulatory Framework for Peptide Characterization
| Regulatory Framework | Applicable Drug Class | Impurity Identification Threshold | Qualification Threshold | Critical Analytical Expectation |
|---|---|---|---|---|
| USFDA ANDA Sameness | Synthetic peptides (≤ 40 amino acids) | ≥ 0.10% of drug substance | ≥ 0.15% or 1.0 mg/day | Direct comparison of the generic product and reference listed drug across multiple batches subjected to identical stress conditions |
| EMA Synthetic Peptide Guideline | Synthetic peptides and peptide-conjugates | ≥ 0.10% reporting threshold | Aligned with Ph. Eur. Monograph 2034 requirements | Comprehensive upstream control strategy beginning with protected amino acids together with stringent control of residual TFA |
| ICH Q1A / Q2 / Q14 | Global harmonization guidelines | Variable according to Maximum Daily Dose (MDD) | Consistent with ICH Q3A(R2) requirements | Fully validated, stability-indicating analytical procedures demonstrating accuracy, robustness, and reproducibility |
Enhance your secondary structure analysis with CD Spectroscopy for Peptide Secondary Structure Characterization.
To satisfy these stringent regulatory expectations, ResolveMass Laboratories Inc., a USFDA-registered pharmaceutical laboratory (FDA Establishment Identifier No. 3042696771), performs comprehensive peptide characterization and ANDA sameness studies. Operating from Canada while serving pharmaceutical organizations throughout North America, the laboratory’s multidisciplinary team of Ph.D.-level scientists employs advanced, multi-technique analytical platforms to generate scientifically defensible and audit-ready data packages supporting IND, NDA, and ANDA submissions.
Need assistance with Cyclic Peptide Characterization? Our experts are here to help.
Conclusion
Peptide Physicochemical Characterization Services provide the comprehensive structural, physicochemical, and thermodynamic evidence necessary to support the development and regulatory approval of modern peptide therapeutics. By integrating orthogonal chromatographic, spectroscopic, and biophysical analytical techniques, these services enable pharmaceutical developers to establish product quality, verify generic sameness, characterize degradation pathways, and satisfy regulatory expectations across global markets.
For generic synthetic peptides, demonstrating physicochemical sameness while maintaining rigorous control of degradation products at the 0.10% reporting threshold is a mandatory requirement for both FDA and EMA regulatory submissions. Collaborating with an experienced analytical contract research organization (CRO) is therefore essential for successfully navigating these complex scientific and regulatory requirements. ResolveMass Laboratories Inc. provides comprehensive end-to-end analytical and regulatory support, including method validation in accordance with ICH Q2 and Q14 guidelines, as well as complete Module 3 Common Technical Document (CTD) Chemistry, Manufacturing, and Controls (CMC) documentation. The company’s extensive expertise in the characterization of complex, modified, and lipidated peptides enables biopharmaceutical developers to reduce regulatory risk while accelerating product development and commercialization timelines.
To learn more about advanced peptide characterization workflows or to begin a collaboration on generic peptide development projects, visit the ResolveMass Laboratories Inc. Contact Page.
Frequently Asked Questions
Trifluoroacetic acid (TFA) is frequently used during solid-phase peptide synthesis (SPPS) and purification as a counter-ion or mobile-phase additive. However, excessive residual TFA may negatively affect peptide stability and can raise toxicological concerns. For this reason, the EMA requires manufacturers to justify its use, establish acceptable residual limits, and demonstrate appropriate control throughout the manufacturing process.
The susceptibility of Asparagine (Asn) to spontaneous deamidation depends greatly on the amino acid positioned immediately adjacent to it, particularly on the C-terminal side. Smaller residues such as Glycine (Gly) and Serine (Ser) promote the formation of the cyclic succinimide intermediate, increasing the reaction rate. In contrast, bulkier amino acids create steric hindrance that slows this degradation pathway and improves peptide stability.
Capillary Zone Electrophoresis (CZE) determines peptide pKa values by measuring changes in effective electrophoretic mobility (μeff) across a carefully controlled pH gradient. As the ionization state of the peptide changes, its migration behavior also changes, allowing accurate identification of dissociation constants. Since CZE separates impurities from the analyte during analysis, it provides reliable results even for samples with lower purity or limited aqueous solubility.
The OECD 123 slow-stirring method is specifically designed for highly hydrophobic and lipidated peptides that are prone to emulsion formation during conventional partition studies. Gentle mixing minimizes the formation of stable n-octanol/water emulsions, allowing accurate equilibrium to be achieved without disturbing the two-phase system. As a result, this method produces more reliable Log P measurements for compounds with very high lipophilicity.
Reversed-phase HPLC (RP-HPLC) provides excellent chromatographic separation of peptides, impurities, and closely related variants based on their physicochemical properties. When coupled with mass spectrometry (LC-MS), the system also delivers accurate molecular weight determination and structural confirmation. This combined analytical approach enables sensitive detection of degradation products, process impurities, and sequence modifications, making it one of the most reliable techniques for peptide purity assessment.
The dissociation constant (pKa) describes the pH at which a specific ionizable functional group within a peptide is equally distributed between its protonated and deprotonated forms. In contrast, the isoelectric point (pI) represents the overall pH at which the peptide carries no net electrical charge. While pKa values describe the behavior of individual ionizable groups, the pI reflects the combined contribution of all charged groups within the entire peptide molecule.
Conversion of Aspartic acid (Asp) to isoaspartic acid (isoAsp) does not produce a measurable change in molecular weight because the modification is isobaric. However, the rearrangement changes the peptide backbone by introducing an additional methylene group, which alters local conformation, surface hydrophobicity, and charge distribution. These structural differences modify the peptide’s interaction with chromatographic stationary phases, producing distinct retention times during chromatographic analysis.
For generic synthetic peptides referencing a recombinant reference listed drug (RLD), the FDA requires identification and characterization of peptide-related impurities present at concentrations of 0.10% or greater within the drug substance. Any impurity that is unique to the generic product must also be scientifically justified and evaluated for potential safety concerns, including immunogenicity. Comprehensive impurity profiling is therefore an essential component of regulatory submissions.
Size-Exclusion Chromatography (SEC-HPLC) is highly effective for quantifying peptide monomers and smaller soluble aggregates, but very large aggregates may not always be accurately detected because they can be retained within the chromatographic system or dissociate during sample preparation. Right Angle Light Scattering (RALS) complements SEC-HPLC by measuring scattered light directly from particles in their native solution without filtration or excessive dilution. Using both techniques together provides a more complete and accurate evaluation of peptide aggregation behavior.
Reference:
- Duncan, K. (2024). CMC regulatory experiences and expectations for peptides [Presentation slides]. United States Pharmacopeia (USP). https://www.usp.org/sites/default/files/usp/document/events-and-training/03-CMC-Regulatory-Experiences-and-Expectations_Katharine-Duncan.pdf
- U.S. Food and Drug Administration. (2021, May). ANDAs for certain highly purified synthetic peptide drug products that refer to listed drugs of rDNA origin: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/107622/download
- Patel, A., & Patel, R. (2024). Analytical techniques for peptide-based drug development: Characterization, stability and quality control. International Journal of Science and Research Archive, 12(1), 3140–3159. https://doi.org/10.30574/ijsra.2024.12.1.1108
- Elsayed, Y. Y., Kühl, T., & Imhof, D. (2025). Regulatory guidelines for the analysis of therapeutic peptides and proteins. Journal of Peptide Science, 31(3), e70001. https://doi.org/10.1002/psc.70001
- Subirats, X., Fuguet, E., Rosés, M., Bosch, E., & Ràfols, C. (2015). Methods for pKa determination (I): Potentiometry, spectrophotometry, and capillary electrophoresis. In J. Reedijk (Ed.), Reference module in chemistry, molecular sciences and chemical engineering. Elsevier. https://doi.org/10.1016/B978-0-12-409547-2.11559-8
- European Medicines Agency. (2018, November 30). Draft guideline on the environmental risk assessment of medicinal products for human use (Revision 1) (EMEA/CHMP/SWP/4447/00 Rev. 1). https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-environmental-risk-assessment-medicinal-products-human-use-revision-1_en.pdf
- U.S. Food and Drug Administration. (2022, November). Sameness evaluations in an ANDA—Active ingredients: Guidance for industry (Draft guidance). U.S. Department of Health and Human Services. https://www.fda.gov/media/163018/download

