
Introduction
The Glass Transition Temperature (Tg) of PLGA is far more than a routine material specification. It functions as the molecular control point that determines the overall behavior and performance of PLGA-based drug delivery systems. When a PLGA matrix is exposed to temperatures at or near its Tg, the polymer chains shift from a rigid, glassy structure into a softer, rubbery state with increased molecular mobility. This transformation in polymer dynamics triggers a cascade of critical changes, including accelerated water uptake, altered erosion kinetics, and enhanced diffusion of drug molecules that were previously confined within a dense polymer network. For scientists developing microspheres, implants, nanoparticles, or in situ forming depots, a detailed understanding of how Tg governs these mechanisms is essential for successful formulation design.
One of the major complexities in PLGA formulation science is that Tg is not a fixed or permanent value. Instead, it is a highly dynamic parameter influenced by polymer composition, molecular weight, processing conditions, residual solvents, moisture uptake, plasticization effects, and environmental exposure. A formulation that appears thermally stable during laboratory storage may behave very differently once exposed to physiological conditions at 37°C. This article explores the mechanistic relationship between PLGA Tg, drug release kinetics, and formulation stability in the level of detail required for advanced formulation development and optimization.
For projects requiring strict adherence to regulatory standards, ensure your starting materials meet international monographs. Learn more by visiting the Pharmaceutical-Grade PLGA Supplier Page.
Share via:
Article Summary:
- PLGA Glass Transition Temperature (Tg) is a key physicochemical property that influences polymer flexibility, drug diffusion, release behavior, and long-term formulation stability.
- The Tg of PLGA generally ranges between approximately 37°C and 60°C, and even small temperature changes can significantly affect release kinetics and degradation behavior.
- Major factors affecting Tg include the lactide:glycolide (LA:GA) ratio, molecular weight, polymer end-group chemistry, residual solvents, plasticizer levels, and drug-polymer interactions.
- High drug loading may reduce Tg through plasticization effects, increasing polymer chain mobility and accelerating drug release rates.
- Storage conditions above or near Tg can destabilize PLGA matrices, resulting in structural relaxation, phase separation, burst release, and shortened shelf life.
- Differential Scanning Calorimetry (DSC) is the most commonly used technique for Tg analysis, while modulated DSC and thermomechanical methods provide additional insight into polymer transition behavior.
- In advanced formulation development, Tg should be intentionally optimized as a formulation design parameter rather than treated solely as a routine characterization value.
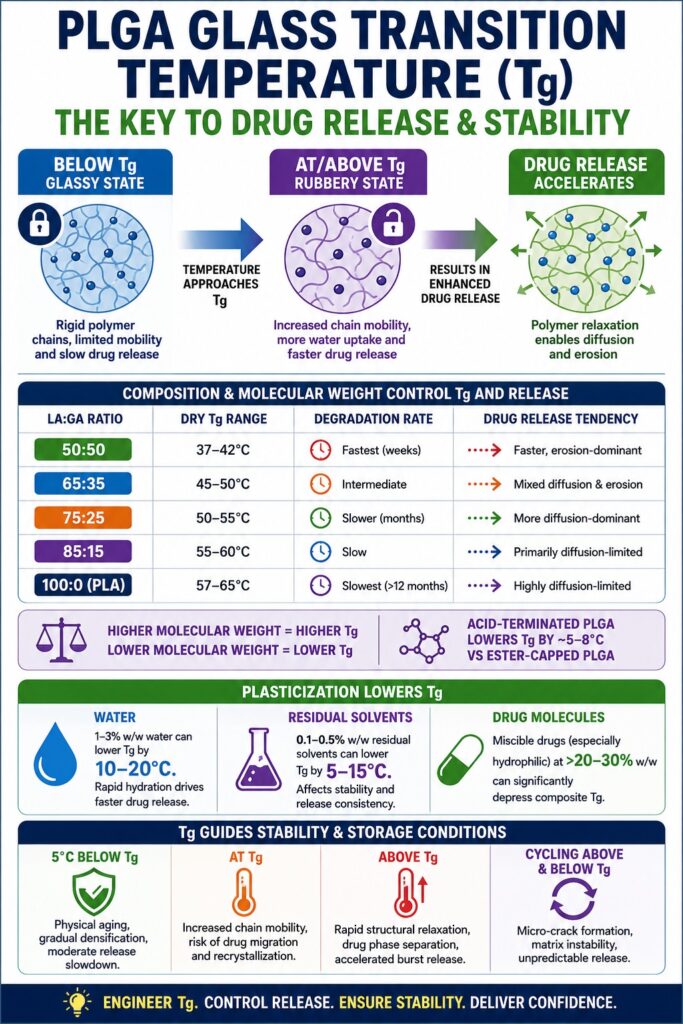
The Mechanistic Relationship Between Tg and Polymer Chain Mobility in PLGA
At temperatures below Tg, PLGA polymer chains exist in a rigid, glassy state characterized by minimal molecular motion. Above Tg, chain segment mobility increases dramatically, directly influencing the diffusion behavior of both water molecules entering the matrix and drug molecules leaving it.
In glassy PLGA systems where the operating temperature is significantly lower than Tg (T << Tg), free volume within the polymer network remains extremely limited. Drug molecules embedded inside the polymer matrix become kinetically trapped because diffusion pathways are highly restricted by the stiffness of the surrounding polymer scaffold. As the temperature approaches and eventually exceeds Tg, the Williams-Landel-Ferry (WLF) model predicts a rapid and highly non-linear increase in polymer chain mobility. This transition is not gradual or mild. Even a temperature increase of only 3–5°C above Tg can raise segmental mobility by several orders of magnitude.
For PLGA-based microspheres and implantable depots, the most relevant operational temperature is physiological body temperature at 37°C. When a formulation possesses a Tg within the range of approximately 38–42°C, the polymer effectively exists near the glassy-to-rubbery transition boundary under in vivo conditions. This creates a highly sensitive release environment in which several important events occur simultaneously:
- Water-induced plasticization can lower the effective Tg below 37°C within only a few hours after hydration.
- Drug release mechanisms shift from being diffusion-limited to matrix-relaxation-controlled.
- Polymer erosion patterns may transition from relatively linear behavior into biphasic or multiphasic release profiles.
The practical consequence is significant. Tg should not be viewed solely as a storage or stability parameter. Instead, it serves as one of the primary kinetic control variables governing release rate engineering in PLGA formulations.
To establish highly predictable degradation pathways for your next depot formulation, explore specialized materials at PLGA for Depot Formulation.
How LA:GA Ratio and Molecular Weight Regulate PLGA Tg and Drug Release Behavior
The lactide-to-glycolide (LA:GA) ratio is one of the most influential parameters controlling the Tg of PLGA. Increasing the lactide content generally raises Tg because the methyl side group present in lactic acid units restricts polymer chain movement and reduces chain flexibility.
The structure-property relationship between PLGA composition and Tg has been thoroughly studied, although it is often applied inconsistently during formulation development. Typical trends are summarized below:
| PLGA Composition (LA:GA) | Approximate Dry Tg Range | Degradation Rate | Drug Release Tendency |
|---|---|---|---|
| 50:50 | 37–42°C | Fastest (weeks) | Faster, erosion-dominant |
| 65:35 | 45–50°C | Intermediate | Mixed diffusion and erosion |
| 75:25 | 50–55°C | Slower (months) | More diffusion-dominant |
| 85:15 | 55–60°C | Slow | Primarily diffusion-limited |
| PLA (100:0) | 57–65°C | Slowest (>12 months) | Highly diffusion-limited |
Molecular weight introduces an additional level of control over Tg:
- Higher molecular weight PLGA increases chain entanglement density, which elevates Tg.
- Lower molecular weight PLGA contains more chain ends and greater free volume, leading to reduced Tg values.
- Reducing molecular weight from approximately 75 kDa to 15 kDa may decrease Tg by as much as 10–15°C, substantially altering in vivo depot performance.
End-group chemistry is another critical but frequently underestimated factor affecting Tg. Acid-terminated PLGA degrades more rapidly than ester-capped PLGA because the free carboxylic acid groups are more hydrophilic and facilitate faster water penetration into the polymer matrix. Under identical molecular weight and LA:GA conditions, acid-terminated PLGA may exhibit an in situ Tg that is approximately 5–8°C lower than the corresponding ester-capped polymer in physiological environments.
If your targeted delivery window requires specific polymer ratios, discover options at PLGA Ratio & Release Kinetics.
If your formulation demands a different aliphatic polyester entirely, check out the alternatives at PCL Supplier & PLA Supplier.
Plasticization Effects: Water, Residual Solvents, and Drug Molecules as Tg Depressants
Plasticization represents one of the most important and underestimated mechanisms responsible for Tg reduction during PLGA formulation development. Water, residual solvents, and incorporated drug molecules can all significantly depress Tg and alter release performance.
a. Water Plasticization
The Gordon-Taylor equation is commonly used to describe Tg depression caused by plasticizer absorption. In PLGA systems, even relatively small amounts of absorbed water, typically around 1–3% w/w, can lower Tg by 10–20°C compared with the dry polymer.
PLGA microspheres exposed to aqueous biological environments begin absorbing water almost immediately after administration. As a result, a formulation with an initial dry Tg of 45°C may experience a reduction in hydrated Tg to approximately 30–35°C within the first 24 hours. Under these conditions, the polymer rapidly enters the rubbery state at body temperature much earlier than originally anticipated.
This phenomenon highlights a critical limitation in conventional thermal analysis. Tg values measured using dry polymer powders by differential scanning calorimetry (DSC) often fail to represent the true operating Tg of the hydrated polymer during in vitro release testing or in vivo administration. Therefore, release studies conducted in aqueous environments must account for progressive water-induced plasticization throughout the release period.
For a deeper analysis of how molecular weight variations dictate matrix hydration and subsequent drug transport, read the full breakdown at PLGA Molecular Weight & Drug Release.
b. Residual Solvent Plasticization
Residual solvents originating from manufacturing processes such as emulsion-solvent evaporation or nanoprecipitation can also act as strong plasticizers. Common solvents including ethyl acetate, methylene chloride, and acetonitrile may significantly depress Tg even when present at very low residual concentrations.
Residual solvent levels as low as 0.1–0.5% w/w may reduce PLGA Tg by approximately 5–15°C. This creates several important formulation concerns:
- Formulations that fail to meet ICH Q3C residual solvent specifications may simultaneously suffer from compromised thermal stability.
- Incomplete solvent removal during drying or lyophilization can introduce batch-to-batch variability in Tg, leading to inconsistent release kinetics despite otherwise identical processing conditions.
c. Drug Molecule Plasticization
Drug-induced plasticization is one of the most formulation-specific Tg modulation mechanisms. When a drug is miscible with PLGA and possesses a Tg lower than that of the polymer, the Fox equation predicts that the composite Tg will fall somewhere between the Tg values of the two individual components.
Several trends are commonly observed:
- Low molecular weight hydrophilic compounds, including peptides, proteins, and water-soluble APIs, often function as stronger plasticizers than hydrophobic drugs.
- Drug loading levels above approximately 20–30% w/w may substantially reduce the composite Tg, sometimes lowering it below the desired storage temperature.
- In less common situations, drugs or co-encapsulated polymers possessing Tg values higher than PLGA may elevate the composite Tg, thereby slowing drug diffusion and reducing sensitivity to plasticization effects.
For long-acting therapeutics requiring precision matrix control to counteract drug-induced plasticization, view available polymer grades at PLGA for Controlled Release.
The Role of PLGA Tg in Formulation Stability and Storage Conditions
Storing PLGA formulations above their Tg accelerates structural relaxation, polymer chain rearrangement, and phase separation processes, all of which directly affect drug distribution uniformity and long-term release predictability.
A glassy polymer exists in a thermodynamically non-equilibrium state. Even when stored below Tg, PLGA gradually undergoes physical aging, which is an entropy-driven process involving densification and free-volume reduction over time. Physical aging tightens polymer chain packing and may lead to:
- Reduced initial burst release
- Altered pore architecture within microspheres
- Changes in implant mechanical properties
However, exposure to temperatures at or above Tg produces substantially more severe consequences.
| Storage Condition | Consequence |
|---|---|
| 5°C below Tg | Physical aging, gradual densification, moderate release slowdown |
| At Tg | Increased chain mobility, risk of drug migration and recrystallization |
| Above Tg | Rapid structural relaxation, drug phase separation, accelerated burst release |
| Cycling above and below Tg | Micro-crack formation, matrix instability, unpredictable release behavior |
These principles explain why PLGA formulations are commonly stored under refrigerated conditions at 2–8°C or even frozen at −20°C. The temperature gap between storage conditions and Tg must be intentionally engineered during formulation design rather than assumed to be adequate.
Measuring PLGA Tg: Analytical Techniques and Their Importance
Differential Scanning Calorimetry (DSC) remains the standard analytical method for measuring PLGA Tg. However, accurate interpretation requires careful experimental design because Tg in PLGA appears as a subtle step change in heat capacity rather than a sharp thermal peak. In physically aged samples, enthalpy relaxation can further obscure the transition.
Recommended Analytical Approaches
Standard DSC
Standard DSC typically employs a first-heating scan at approximately 10–20°C/min. Tg is identified as the midpoint of the heat-capacity inflection. To eliminate artifacts caused by previous thermal history, the first heating cycle should be followed by controlled quenching and a second-heating scan.
Modulated DSC (mDSC or MDSC)
Modulated DSC separates reversing thermal events such as Tg from non-reversing events including enthalpy relaxation and crystallization. This technique is especially valuable for aged PLGA samples where enthalpy relaxation peaks may distort or shift the apparent Tg by several degrees.
Dynamic Mechanical Analysis (DMA)
DMA evaluates storage modulus (E’) and loss tangent (tan δ) as functions of temperature. Because DMA is highly sensitive to molecular mobility transitions, it is particularly useful for characterizing PLGA films, implants, and mechanically relevant dosage forms.
Thermogravimetric Analysis (TGA) Coupled with DSC
Combining TGA with DSC helps confirm whether apparent Tg reductions are truly caused by polymer transitions or are instead artifacts resulting from moisture evaporation or residual solvent loss during heating.
A critical analytical mistake in formulation development is measuring Tg only on raw PLGA pellets or unprocessed polymer powder. Processing steps such as emulsification, solvent exposure, drug loading, drying, and lyophilization frequently alter the final Tg substantially. Therefore, Tg should always be measured on the finished formulation or on a processing-equivalent surrogate sample.
When transition profiles necessitate highly specific macromolecular tuning, you can review custom options via Custom PLGA Synthesis.
Engineering Tg as a Primary Formulation Design Parameter
Modern PLGA formulation development increasingly treats Tg as a deliberate design target rather than merely a characterization result obtained after formulation completion. Advanced formulation teams now engineer Tg intentionally by selecting appropriate polymer grades, drug loadings, and excipient systems to achieve a desired thermal operating window.
A practical framework for Tg engineering in PLGA depot systems includes the following steps:
- Define the intended operating temperature range, including physiological exposure at 37°C and long-term storage conditions such as 2–8°C or −20°C.
- Establish a minimum dry Tg target, commonly ≥50°C, to maintain an adequate safety margin above physiological temperature after accounting for water-induced plasticization.
- Select an appropriate polymer grade using LA:GA ratio, molecular weight, and end-group chemistry to achieve the target Tg range.
- Predict composite Tg behavior using equations such as the Fox or Gordon-Taylor models at the intended drug loading.
- Evaluate residual moisture and solvent content carefully to identify plasticizer-related Tg depression after drying or lyophilization.
- Correlate Tg with in vitro and in vivo release performance to confirm alignment with the intended sustained-release duration, such as one-month, three-month, or six-month release profiles.
At ResolveMass Laboratories Inc., this design-of-experiments (DoE) strategy for Tg engineering, incorporating polymer selection, drug characterization, and thermal analysis, is routinely applied in advanced PLGA formulation development programs.
For organizations planning to scale up their optimized formulations under rigorous regulatory oversight, secure reliable infrastructure by visiting PLGA Contract Manufacturing.
Conclusion
The Glass Transition Temperature of PLGA serves as the molecular fulcrum that balances drug release kinetics and formulation stability. Recognizing that Tg is not a static material constant, but rather a dynamic and environmentally responsive property, is essential for rational formulation development. Its value continuously changes in response to polymer composition, molecular weight, hydration, residual solvents, and drug-polymer interactions.
Formulation scientists who systematically engineer Tg, account for in situ plasticization effects, and validate thermal behavior under both physiological and storage-relevant conditions gain significantly greater control over release performance and formulation reproducibility. In contrast, approaches that overlook Tg dynamics often result in inconsistent release profiles and reduced long-term stability.
From PLGA microspheres to long-acting injectable depots and implantable delivery systems, mastering the principles governing Glass Transition Temperature is fundamental to developing formulations that are stable, predictable, scalable, and clinically translatable.
Frequently Asked Questions (FAQs)
When PLGA is exposed to physiological fluids, the polymer rapidly absorbs water from the surrounding environment. Water molecules act as plasticizers and reduce the Tg of the polymer by increasing chain flexibility and free volume. In many formulations, the hydrated Tg can decrease by approximately 10–20°C compared with the dry state. As a result, PLGA may transition into a rubbery state at body temperature, leading to faster drug diffusion, accelerated erosion, and altered release kinetics.
The faster release behavior of 50:50 PLGA is mainly linked to its lower Tg, greater hydrophilicity, and faster hydrolytic degradation compared with 75:25 PLGA. Since its Tg is closer to physiological temperature, polymer chain mobility increases more easily under in vivo conditions. This enhanced mobility promotes quicker water penetration and polymer erosion. Consequently, both drug diffusion and matrix degradation occur at a significantly faster rate.
Yes, crystallization can interfere with accurate Tg determination in PLGA systems. Although PLGA is typically considered an amorphous polymer, formulations with high lactide content may develop crystalline regions under certain thermal conditions. The formation of crystalline domains reduces the amount of amorphous material available for glass transition detection, making the Tg signal appear weaker during DSC analysis. In some cases, drug-induced crystallization may also complicate thermal characterization and lead to misleading Tg values.
Lyophilization generally increases the Tg of PLGA formulations because the drying process removes residual water and organic solvents that normally act as plasticizers. However, the final effect depends heavily on formulation composition. Cryoprotectants such as trehalose, sucrose, or mannitol may remain within the polymer matrix after freeze-drying and can lower Tg if they interact with the polymer chains. Therefore, the overall Tg shift after lyophilization depends on both solvent removal efficiency and excipient compatibility.
Tg plays a major role in determining how closely in vitro release data reflect actual in vivo performance. During in vitro testing, PLGA formulations are commonly exposed to aqueous buffers at 37°C, conditions that often place the polymer near or above its hydrated Tg. In vivo environments, however, involve additional factors such as tissue interactions, protein adsorption, pH variations, and mechanical stress, all of which can influence polymer mobility and degradation behavior. Incorporating Tg-related variables into IVIVC models improves the predictive accuracy of drug release performance.
Drug recrystallization within PLGA matrices is strongly influenced by polymer chain mobility, which increases significantly above Tg. When formulations are stored at temperatures exceeding Tg, drug molecules gain sufficient mobility to migrate and form crystalline structures over time. This transformation can alter release kinetics, reduce dose uniformity, and compromise formulation stability. Maintaining storage temperatures below Tg helps preserve the amorphous dispersion state and minimizes the risk of recrystallization.
There is no universal Tg specification that applies to all PLGA injectable systems because each formulation has unique composition and release requirements. However, many formulation scientists aim for a dry Tg in the range of approximately 45–55°C to ensure adequate thermal stability during refrigerated storage and physiological exposure. Since hydration can substantially reduce Tg after administration, a sufficient safety margin must always be built into the formulation design. Final Tg targets should be validated experimentally rather than assumed theoretically.
Combining PLGA with polymers such as PEG or polycaprolactone can significantly modify both Tg and release behavior. Low-Tg polymers increase chain flexibility and typically reduce the composite Tg of the blend, which enhances water uptake and accelerates drug diffusion. In miscible systems, the final Tg usually follows predictive blending models such as the Fox equation. In immiscible systems, separate polymer phases may form, each displaying distinct Tg values and independent release characteristics.
Reference:
- Makadia, H. K., & Siegel, S. J. (2011). Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers, 3(3), 1377–1397. https://doi.org/10.3390/polym3031377
- Fredenberg, S., Wahlgren, M., Reslow, M., & Axelsson, A. (2011). The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. International Journal of Pharmaceutics, 415(1–2), 34–52. https://doi.org/10.1016/j.ijpharm.2011.05.049
- Gentile, P., Chiono, V., Carmagnola, I., & Hatton, P. V. (2014). An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. International Journal of Molecular Sciences, 15(3), 3640–3659. https://doi.org/10.3390/ijms15033640
- Sill, T. J., & von Recum, H. A. (2008). Electrospinning: Applications in drug delivery and tissue engineering. Biomaterials, 29(13), 1989–2006. https://doi.org/10.1016/j.biomaterials.2008.01.011
- Corrigan, O. I., & Li, X. (2009). Quantifying drug release from PLGA nanoparticulates. European Journal of Pharmaceutical Sciences, 37(3–4), 477–485. https://doi.org/10.1016/j.ejps.2009.04.004

