
Introduction
Stability testing in bioanalysis, as outlined in the ICH M10 guideline, provides scientific evidence that every stage of sample collection, preparation, processing, and storage preserves the integrity of analyte concentrations. Before the adoption of this harmonized guideline, pharmaceutical companies had to navigate a complex network of region-specific regulatory requirements, addressing varying expectations established by the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and numerous other national regulatory authorities. This lack of harmonization frequently resulted in duplicated validation efforts, extended regulatory review timelines, and an increased risk of compliance issues, particularly during global, multi-center clinical studies.
Learn more about the latest regulatory frameworks in our guide on Bioanalytical Strategy in Drug Development
The adoption and implementation of the ICH M10 guideline for Bioanalytical Method Validation (BMV) and Study Sample Analysis introduced a unified scientific framework that simplifies global drug development while ensuring the reliability of pharmacokinetic (PK) and toxicokinetic (TK) data. For contract research organizations (CROs) as well as pharmaceutical and biotechnology companies, establishing comprehensive procedures for stability testing in bioanalysis has evolved from being a regional regulatory expectation into a fundamental requirement for obtaining international regulatory approval.
Ensure your laboratory protocols meet current standards by reviewing our ICH M10 Bioanalytical Method Validation Guidelines
To satisfy these globally accepted standards, modern contract laboratories utilize advanced analytical technologies, including liquid chromatography-tandem mass spectrometry (LC-MS/MS) and high-resolution nuclear magnetic resonance (NMR) instrumentation, to assess the molecular integrity of small molecules, peptides, oligonucleotides, and macromolecular therapeutics within complex biological matrices. Adherence to these stringent scientific requirements ensures that the data submitted to regulatory agencies accurately represent the true in vivo concentrations of the parent drug and its metabolites at the exact time of sample collection.
Discover how advanced technology enhances precision in our Enabling Bioanalytical Studies overview
Share via:
Article Summary:
- ICH M10 establishes a globally harmonized framework for bioanalytical stability testing, ensuring that biological samples maintain analyte integrity throughout collection, processing, storage, and analysis while supporting consistent regulatory compliance worldwide.
- Freeze-thaw stability testing confirms analyte reliability after repeated freezing and thawing, requiring at least three complete cycles, separate QC aliquots, appropriate storage temperatures, and a minimum 12-hour freezing period to simulate real laboratory conditions.
- Bench-top stability testing evaluates analyte performance during routine sample handling by validating uninterrupted exposure times under laboratory conditions and addressing potential risks such as enzymatic degradation, adsorption, photodegradation, and temperature-related changes.
- Long-term stability studies demonstrate that analytes remain stable throughout the intended frozen storage period, ensuring that measured concentrations accurately represent the original biological sample even after extended storage before analysis.
- The ICH M10 guideline introduces efficient validation strategies, including scientifically justified bracketing across storage temperatures and the use of dilution quality control (DIQC) samples for studies involving analyte concentrations above the validated calibration range.
- Validation requirements differ between chromatographic methods (LC-MS/MS) and ligand binding assays (LBA) because each platform analyzes different therapeutic molecules and therefore follows distinct quality control levels, acceptance criteria, and stability assessment procedures.
- Comprehensive stability testing strengthens data integrity and regulatory confidence, ensuring that pharmacokinetic and toxicokinetic results remain accurate, reproducible, and suitable for global drug development and regulatory submissions.
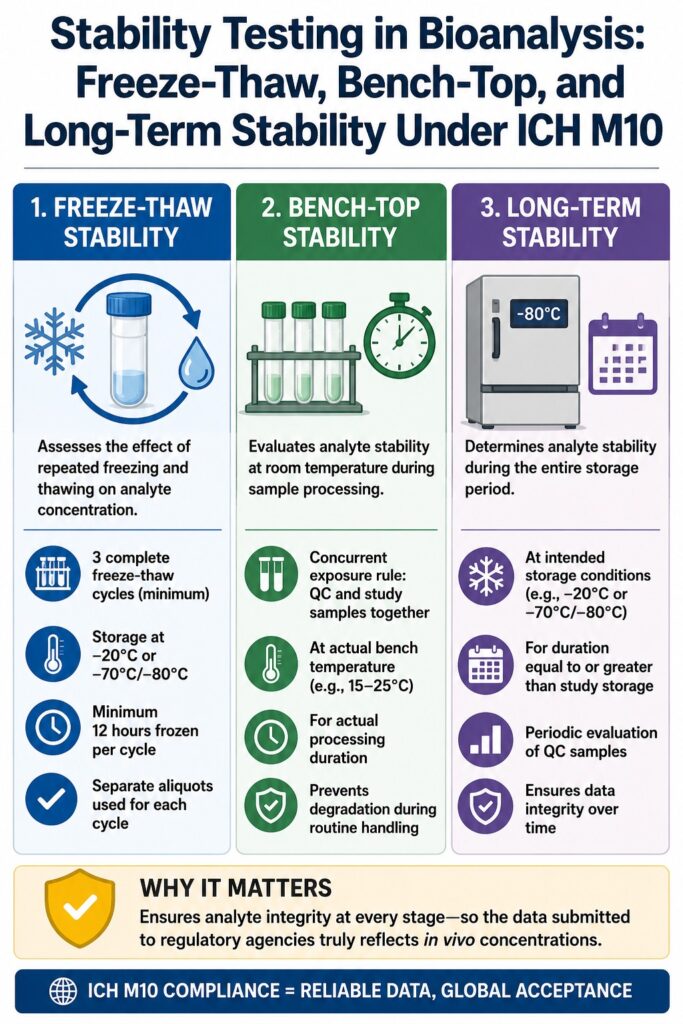
Scientific Rigor in Freeze-Thaw Stability Testing in Bioanalysis
Freeze-thaw stability testing in bioanalysis assesses the physical and chemical effects associated with repeatedly removing biological samples from frozen storage by evaluating quality control (QC) samples through a minimum of three complete freeze-thaw cycles. The primary scientific objective of this study is to determine whether temperature transitions, ice crystal formation, or cryo-concentration introduce measurable changes in analyte concentrations when clinical samples undergo repeated thawing for initial analysis, dilution, or subsequent reanalysis.
The thermodynamic stress generated during repeated freezing and thawing can induce significant physicochemical changes within biological matrices. Therefore, the ICH M10 guideline establishes specific operational requirements that laboratories must follow to minimize variability and maintain data integrity.
Bulk QC Preparation and Aliquoting: To obtain representative and reproducible stability data, the guideline requires laboratories to prepare a single bulk QC sample at both low and high concentration levels. This bulk preparation must then be divided into at least three independent aliquots, with each aliquot stored, subjected to stress conditions, and analyzed separately. Evaluating individual aliquots rather than repeatedly accessing the same sample tube minimizes localized analyte migration, commonly referred to as the freeze-concentration effect, thereby providing a more accurate simulation of actual clinical sample handling.
Freezing Temperature Profiles: The storage temperature used throughout the freezing phase must accurately reflect the intended storage conditions for study samples, typically −20°C or −70°C/−80°C. For small-molecule analytes, validation at −20°C is generally considered sufficient when samples are exclusively stored under those conditions. In contrast, biological therapeutics and macromolecular compounds require stability evaluations at both −20°C and −70°C/−80°C whenever samples are expected to be stored under both temperature conditions during the study.
Strict Timing and Solidification: Each freeze-thaw cycle must include a minimum frozen storage period of 12 hours before the sample is thawed again. This minimum storage interval allows complete thermal equilibration and consistent crystallization of the biological matrix, effectively reproducing the structural changes that occur in frozen clinical samples during long-term storage.
Procedural Replication: The thawing procedure performed during validation must accurately replicate the laboratory’s routine operating procedures for clinical study samples, whether thawing is conducted on wet ice, at ambient room temperature, or using controlled water baths. Furthermore, the total number of validated freeze-thaw cycles must be equal to or greater than the maximum number of cycles expected during actual sample handling, with a minimum validation requirement of three complete freeze-thaw cycles.
Avoid common errors by reading our breakdown of Common Bioanalytical Mistakes
Operational Protocols for Bench-Top Stability Testing in Bioanalysis
Bench-top stability testing in bioanalysis evaluates the stability of an analyte within a biological matrix under the exact temperature and exposure duration encountered during routine laboratory processing. This assessment is particularly important because analytes may undergo enzymatic hydrolysis, oxidation, photolysis, or adsorption onto laboratory surfaces when biological specimens remain thawed on the laboratory bench during sample preparation and extraction procedures.
To minimize these biochemical risks, the ICH M10 guideline establishes strict operational requirements governing bench-top stability assessments.
The Concurrent Exposure Rule
To eliminate the possibility of overestimating analyte stability, the ICH M10 guideline enforces the concurrent exposure rule. Under previous regulatory practices, some laboratories estimated total bench-top stability by combining multiple short periods during which samples remained at room temperature across several freeze-thaw events. This cumulative calculation method is no longer considered acceptable under the ICH M10 framework.
Instead, the validated bench-top stability period must represent one uninterrupted exposure that encompasses the complete laboratory processing workflow, beginning with initial sample thawing and vortex mixing and continuing through extraction, dilution, and the final transfer of prepared samples to the analytical instrument.
Multi-Drug Combinations and Fixed-Dose Regimens
For clinical studies involving fixed-dose combinations or specifically designated multidrug treatment regimens, bench-top stability evaluations must be performed using a biological matrix co-spiked with every administered compound. The spiked concentrations should accurately reflect the circulating drug levels expected in patient samples, including the maximum observed concentration (Cₘₐₓ).
Conducting these evaluations is essential for identifying potential chemical interactions among co-administered drugs, competitive enzymatic degradation, or matrix suppression effects that may negatively affect analytical accuracy and compromise the reliability of bioanalytical data.
Validation Strategies for Stress Conditions
To maintain compliance with the ICH M10 guideline, laboratories must establish appropriate validation procedures to address the various stress conditions that biological samples may encounter during bench-top processing. The following table summarizes common stress conditions, their potential effects on the biological matrix, and the recommended validation and mitigation strategies.
| Potential Stress Condition | Matrix-Level Impact | Validation and Mitigation Strategy Under ICH M10 |
|---|---|---|
| Enzymatic Degradation | Rapid enzymatic cleavage of prodrugs, ester-containing compounds, or peptide therapeutics by active plasma enzymes. | Validate the addition of appropriate enzyme inhibitors (for example, esterase inhibitors) to the biological matrix and maintain carefully controlled pH conditions throughout sample preparation using validated diluents. |
| Adsorption to Surfaces | Non-specific adsorption of hydrophobic analytes, including oligonucleotides, onto polypropylene or glass laboratory containers. | Validate the use of low-binding plastic consumables or pre-conditioned container surfaces, particularly when analyzing samples containing analyte concentrations close to the lower limit of quantification (LLOQ). |
| Photolysis / Light Sensitivity | Ultraviolet (UV) light-induced degradation or structural isomerization of photosensitive compounds. | Perform validation under controlled lighting conditions, such as gold fluorescent or amber illumination, utilize amber-colored sample containers, and document all light-protection procedures within the analytical method. |
| Temperature Drift | Accelerated hydrolysis, deconjugation, or other temperature-dependent chemical degradation occurring at elevated ambient temperatures (22°C to 25°C). | Validate sample handling under wet ice conditions (4°C) and establish a scientifically justified maximum continuous bench-top exposure time for routine laboratory operations. |
Improve your laboratory efficiency with our Integrated Chemistry and Bioanalytical CRO solutions
Bracketing and Concentration Thresholds in Long-Term Stability Testing in Bioanalysis
Long-term stability testing in bioanalysis demonstrates that an analyte maintains its chemical integrity during frozen storage for a period equal to or longer than the anticipated storage duration of study samples. This assessment is particularly important because clinical specimens are frequently stored in ultra-low-temperature freezers for several months or even years before bioanalytical testing or regulatory submission. Confirming long-term stability ensures that measured analyte concentrations accurately reflect the original sample composition throughout the entire storage period.
To maximize scientific efficiency while maintaining rigorous analytical standards, the ICH M10 guideline incorporates standardized bracketing approaches and concentration-based validation strategies for long-term stability assessments.
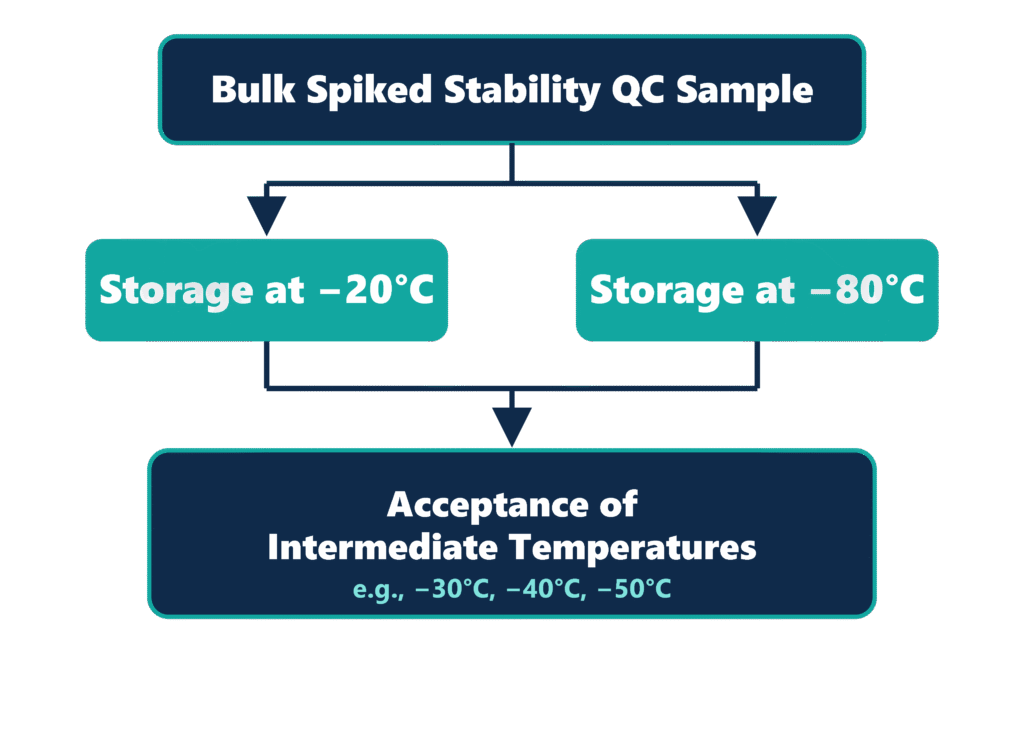
The Bracketing Principle for Biological Therapeutics
For large-molecule biological therapeutics, the ICH M10 guideline permits the application of a scientifically justified bracketing approach across different frozen storage temperatures. When long-term stability has been successfully demonstrated at both −20°C and −70°C/−80°C, the analyte is considered scientifically stable across all intermediate storage temperatures. Consequently, separate validation studies at intermediate temperatures, such as −30°C or −40°C, are generally unnecessary. This scientifically supported approach reduces redundant validation efforts while simplifying global bioanalytical validation programs.
High Concentration QC Adjustments
When study sample concentrations consistently exceed the validated upper limit of quantification (ULOQ), the concentration of the high stability QC sample should be increased to adequately represent these elevated analyte levels. This situation commonly arises during clinical studies in which maximum plasma concentrations (Cₘₐₓ) exceed the validated calibration range, requiring sample dilution before analysis.
To preserve analytical reliability under these circumstances, laboratories should perform stability assessments using dilution quality control (DIQC) samples. These studies verify that high-concentration biological matrices remain stable during prolonged frozen storage without experiencing molecular aggregation, precipitation, or analyte adsorption to container surfaces that could compromise quantitative accuracy.
Learn how to maintain high standards for your data in our resource on Data Integrity in Bioanalytical Studies
Cross-Platform Stability Parameters: Chromatography vs. Ligand Binding Assays in Stability Testing in Bioanalysis
The practical implementation of stability testing in bioanalysis differs considerably between chromatographic methods and ligand binding assays because each analytical platform is designed to evaluate different classes of therapeutic molecules. Chromatographic methods, most commonly coupled with LC-MS/MS instrumentation, are primarily used for the quantitative analysis of small molecules, peptides, and metabolites. In contrast, ligand binding assays (LBAs) rely on highly specific antibody-antigen interactions to quantify large-molecule biologics, including monoclonal antibodies, biosimilars, and other protein-based therapeutics.
These fundamental analytical differences result in distinct validation procedures, sample preparation requirements, and acceptance criteria under the ICH M10 guideline. The comparison below summarizes the primary validation parameters described for chromatographic methods (Section 3.2) and ligand binding assays (Section 4.2).
| Methodological Metric | Chromatographic Methods (LC-MS/MS) | Ligand Binding Assays (LBA) |
|---|---|---|
| Minimum QC Levels | Four QC levels: LLOQ, Low QC (≤ 3 × LLOQ), Mid QC (30%–50% of the calibration range), and High QC (≥ 75% of the ULOQ). | Five QC levels: LLOQ, Low QC (near the LLOQ), Mid QC (geometric mean of the calibration range), High QC (near the ULOQ), and ULOQ. |
| Stability QC Preparation | One bulk QC sample divided into at least three independent aliquots for each concentration level. | One bulk QC sample prepared at each concentration level, with macromolecular samples frozen for a minimum of 12 hours between freeze-thaw cycles. |
| Acceptance Criteria (Bias and CV) | Mean concentration must remain within ±15% of nominal values (±20% at the LLOQ), with a coefficient of variation (CV) ≤15% (≤20% at the LLOQ). | Mean concentration must remain within ±20% of nominal values (±25% at the LLOQ and ULOQ), with a CV ≤20% (≤25% at the LLOQ and ULOQ). |
| Whole Blood Stability | Mandatory validation using fresh, non-coagulated whole blood whenever plasma serves as the analytical matrix. | Generally not required when plasma or serum stability has already been demonstrated under equivalent storage conditions. |
| Reinjection Reproducibility | Explicitly required to establish the stability and suitability of processed extracts for delayed reinjection. | Not applicable because plate-based ligand binding assays are typically analyzed only once during each assay cycle. |
| Selectivity Lots Required | At least six individual blank matrix sources, along with one hemolyzed and one lipemic matrix source. | At least ten individual blank matrix sources evaluated at both the LLOQ and high QC concentration levels. |
Understand the technical nuances in our article on Robust Bioanalytical Data
Processed Sample Stability versus Reinjection Reproducibility in Chromatographic Systems
Within the ICH M10 framework, processed sample stability and reinjection reproducibility are evaluated as two independent validation parameters. Processed sample stability demonstrates the chemical stability of prepared sample extracts after extraction, whereas reinjection reproducibility confirms that reliable analytical results can still be obtained following unexpected operational delays or instrument interruptions.
Because chromatographic analyses are performed using automated analytical systems, prepared extracts often remain inside temperature-controlled autosampler trays for several hours or, in some cases, several days before injection. If an analytical sequence is interrupted because of an autosampler malfunction, elevated column backpressure, power failure, or another instrument-related issue, the laboratory must have previously validated recovery procedures that allow the analytical run to be resumed without compromising data quality. A clear understanding of the distinction between processed sample stability and reinjection reproducibility is therefore essential for maintaining analytical integrity and ensuring successful recovery of interrupted chromatographic runs.
Processed Sample Stability
Processed sample stability evaluates the length of time extracted samples can remain stored before instrumental analysis without experiencing chemical degradation, solvent evaporation, precipitation, or any other change that could affect analyte quantification. During validation, processed quality control (QC) samples at both low and high concentration levels, with a minimum of three replicates per level, are stored under predefined conditions, such as in an autosampler maintained at 6°C for 48 hours. Following the designated storage period, these samples are analyzed using a freshly prepared calibration curve to determine whether analyte integrity has been maintained.
To satisfy the acceptance criteria established by ICH M10, the mean measured concentrations of the stored QC samples must remain within ±15% of their nominal concentrations. Meeting this requirement demonstrates that the specified storage conditions do not adversely affect the stability of the extracted analytes and confirms the suitability of delayed sample analysis.
Reinjection Reproducibility
Reinjection reproducibility determines whether previously processed sample extracts can be reanalyzed after an unexpected interruption in the analytical sequence while still producing reliable and acceptable results. This validation becomes particularly important when instrument failures, autosampler interruptions, power outages, or other operational issues prevent completion of an analytical batch.
To perform this assessment, an entire analytical run containing the calibration standards together with a minimum of five replicates each of low, medium, and high QC samples is initially analyzed. The complete plate is then stored under validated conditions, typically within the autosampler or under refrigerated storage. After the predetermined storage interval, the entire analytical sequence is reinjected without preparing a new calibration curve. The reinjected QC samples must satisfy the standard batch acceptance criteria, with at least two-thirds of all QC samples and a minimum of 50% of the QC samples at each concentration level remaining within ±15% of their nominal concentrations. Successful completion of this evaluation confirms that analytical runs can be recovered without compromising data integrity.
See how we handle complex analytical needs in our Tissue and CSF Bioanalytical Services
Solution and Matrix Stability Standards under ICH M10
Solution stability testing under the ICH M10 guideline requires independent confirmation of the stability of both the analyte and the internal standard (IS) in stock and working solutions under the storage conditions used during routine laboratory operations. A manufacturer’s Certificate of Analysis (CoA) for the neat reference material alone is not considered sufficient evidence of solution stability. As specified in Section 3.2.8.3 of the guideline, the stability of the analyte and the internal standard in solution must be established independently from the stability of the neat compound.
This requirement ensures that variables such as solvent composition, storage container materials, storage duration, and analyte concentration do not introduce degradation, precipitation, or other physicochemical changes that could affect analytical performance throughout the study.
Verification of Primary Stock Solutions
When preparing primary stock solutions, laboratories are required to verify the accuracy and consistency of solution preparation. This verification is accomplished by independently preparing two separate stock solutions and comparing their detector responses. The percentage difference between the two measured responses must not exceed 5%, as determined using the following equation:
Difference = (|Stock 1 − Stock 2| ÷ Mean Value) × 100 ≤ 5%
Compliance with this acceptance criterion confirms that stock solution preparation is reproducible and that subsequent analytical measurements are based on accurately prepared reference materials.
Internal Standard Stability Requirements
The stability requirements for internal standards under ICH M10 depend on the type of internal standard selected for the analytical method, with different validation expectations applying to stable isotopically labeled compounds and structural analogue compounds.
Stable Isotopically Labeled (SIL) Internal Standard: When a stable isotopically labeled internal standard is used under identical solvent compositions and storage conditions as the analyte, and there is no evidence of isotopic exchange, an independent stability study for the internal standard is generally not required. During method validation, laboratories should confirm isotopic stability by evaluating zero samples to demonstrate the absence of analytical interference or isotopic instability.
Structural Analogue Internal Standard: When a structural analogue is used instead of a stable isotopically labeled compound, both short-term and long-term solution stability studies must be performed using appropriate bracketing concentrations. This additional validation is necessary because structural analogues may differ from the target analyte in extraction recovery, matrix effects, and susceptibility to chemical degradation during storage, making independent stability verification essential.
Optimize your project timelines by exploring our Bioanalytical Services for Rapid Proof of Concept
Unified Global Consensus in Stability Testing in Bioanalysis
The implementation of standardized procedures for stability testing in bioanalysis enables pharmaceutical development programs to generate highly reproducible pharmacokinetic data while satisfying globally harmonized regulatory expectations. The introduction of the ICH M10 guideline represents a significant advancement in international bioanalytical science by aligning validation requirements across major regulatory authorities, including the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), Health Canada, and other global health agencies. This harmonized approach minimizes redundant validation activities, improves regulatory consistency, and facilitates more efficient worldwide drug development and submission processes.
Partner with us for your next program—learn more about our Bioanalytical CRO Partnership
For increasingly complex therapeutic products, including small molecules, peptides, oligonucleotides, monoclonal antibodies, and other biologics, collaboration with an experienced contract laboratory plays a critical role in maintaining regulatory compliance and generating reliable analytical data. Modern bioanalytical testing depends on sophisticated technologies such as high-performance chromatography, advanced mass spectrometry, and high-resolution NMR to evaluate matrix effects, characterize unstable metabolites, and validate high-concentration sample dilution strategies with precision.
Need support for upcoming clinical trials? Explore our Bioanalytical CRO for First-in-Human Studies
For organizations requiring customized study designs, advanced polymer characterization, or fully validated bioanalytical support, partnering with experienced scientific specialists can help ensure that analytical studies are performed in accordance with ICH M10 requirements while delivering accurate, reproducible, and regulatory-compliant results throughout the drug development process.
Let’s connect – Contact us.
Frequently Asked Questions (FAQs)
According to ICH M10, quality control (QC) samples used for freeze-thaw stability assessments must remain completely frozen for a minimum of 12 hours before each thawing cycle. This holding period allows the samples to achieve complete thermal equilibrium and consistent crystal formation within the biological matrix. Following this approach helps reproduce the storage conditions experienced by actual clinical specimens and provides a reliable assessment of analyte stability after repeated freezing and thawing.
The concurrent exposure rule requires bench-top stability to be validated using one uninterrupted period that reflects the complete duration of routine laboratory sample processing. Laboratories are not permitted to estimate stability by combining multiple short room-temperature exposures from separate freeze-thaw events. This requirement ensures that stability data accurately represent the real handling conditions encountered during extraction, preparation, and analysis of study samples.
The high quality control (QC) concentration should be increased whenever the concentrations of study samples consistently exceed the validated upper limit of quantification (ULOQ). This situation is commonly encountered in studies where peak drug concentrations are substantially higher than the established calibration range. Adjusting the high QC level ensures that stability assessments accurately represent the concentration range expected during routine sample analysis.
Processed sample stability evaluates whether extracted analytes remain chemically stable after sample preparation and during storage before analysis. Reinjection reproducibility, on the other hand, confirms that previously prepared extracts can be reinjected after an unexpected analytical interruption without compromising data quality. Although both studies support analytical reliability, they assess different aspects of method performance and are validated independently under ICH M10.
In most cases, whole blood stability testing is not required for large biological molecules if adequate stability has already been established in plasma or serum under the same storage and handling conditions. However, additional whole blood stability studies may become necessary when there is evidence that the analyte interacts with blood cells or undergoes significant changes before plasma separation. The requirement should therefore be based on the specific characteristics of the therapeutic molecule.
ICH M10 recommends performing stock and working solution stability studies using a minimum of three independent replicates. The evaluation should also include a comparison of responses obtained from two separately prepared stock solutions to verify preparation accuracy. The observed difference between these independently prepared solutions should not exceed 5%, demonstrating acceptable consistency and reproducibility.
Dilution QC stability studies are not universally mandatory, but they are strongly recommended whenever sample dilution is expected during routine analysis. Their importance increases in studies such as bioavailability (BA) and bioequivalence (BE) investigations, where analyte concentrations frequently exceed the calibration range. Conducting these evaluations helps confirm that dilution procedures do not introduce instability or affect analytical accuracy.
For fixed-dose combination therapies, stability studies should be conducted using biological matrices spiked with all co-administered analytes included in the treatment regimen. The spiked concentrations should closely represent the expected circulating drug levels, including the maximum concentration (Cₘₐₓ). This approach allows laboratories to assess potential interactions between analytes and verify that the combined presence of multiple compounds does not compromise analytical performance or stability.
Reference:
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2022). ICH M10: Bioanalytical method validation and study sample analysis (Step 4 presentation). https://database.ich.org/sites/default/files/ICH_M10_Step_4_Presentation_2022_1123.pdf
- U.S. Food and Drug Administration. (2022). M10 bioanalytical method validation and study sample analysis: Guidance for industry. https://www.fda.gov/media/167335/download
- AAPS Workshop Report on ICH M10 Bioanalytical Method Validation. (2019). The AAPS Journal, 21(6), Article 111. https://pmc.ncbi.nlm.nih.gov/articles/PMC6904406/
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2024, January 27). ICH M10: Bioanalytical method validation and study sample analysis—Training material. https://database.ich.org/sites/default/files/ICH_M10_EWG_Training_Material_2024_0127.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2019, March 20). ICH M10: Bioanalytical method validation—Step 2 presentation. https://database.ich.org/sites/default/files/M10_EWG_Step2_Presentation.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2022, May 24). ICH harmonised guideline M10: Bioanalytical method validation and study sample analysis. https://database.ich.org/sites/default/files/M10_Guideline_Step4_2022_0524.pdf
- European Medicines Agency. (2023, January 13). ICH guideline M10 on bioanalytical method validation and study sample analysis: Frequently asked questions (FAQ) (EMA/CHMP/ICH/660315/2022). https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-and-study-sample-analysis-frequently-asked-questions-faq_en.pdf

