
Introduction:
A bioanalytical method validation failure is one of the most operationally significant and regulatorily sensitive events in drug development. Despite its importance, it remains insufficiently explored from a practical perspective. Scientific literature thoroughly explains how to validate a method, which acceptance criteria must be satisfied, and which regulatory guidelines apply. However, what laboratories and sponsors often need most is detailed guidance on what happens after a validation failure occurs: what documentation must be completed within the first 24 to 48 hours, which technical investigations are mandatory, how to determine whether partial revalidation is justified or whether a complete revalidation is necessary, and what the regulatory consequences may be at different stages of development.
This article addresses those questions directly and systematically, using a practical, step-by-step approach with reference to ICH M10 (adopted by the FDA in November 2022), the FDA Bioanalytical Method Validation Guidance (May 2018), and published bioanalytical literature discussing ISR failure trends, matrix effect mechanisms, and method recovery outcomes.
Explore Comprehensive Support Options: Learn how specialized partners deliver tailored IND Bioanalytical Support to prevent early-stage validation pitfalls before they disrupt your timelines.
Quick Summary:
- A failure in bioanalytical method validation does not necessarily mean that an entire study becomes unusable. The outcome largely depends on how efficiently the issue is investigated, documented, and resolved through an appropriate corrective strategy.
- The first response to a validation failure should include formal rejection of the analytical run, immediate initiation of a documented deviation, and a structured root cause investigation supported by proper quality records.
- The majority of validation failures generally arise from four major problem areas: QC or calibration acceptance failures, matrix-related interference, analyte stability concerns, and ISR (Incurred Sample Reanalysis) reproducibility issues.
- Based on the nature and severity of the failure, recovery may involve analytical method optimization, targeted partial revalidation, or complete method revalidation in accordance with ICH M10 and FDA 2018 bioanalytical guidance requirements.
- During a validation failure event, regulatory compliance is equally as important as technical troubleshooting. Accurate reporting, adherence to SOP-defined procedures, sponsor communication, and documentation controls must all proceed simultaneously.
- Validation failures occurring in pivotal or late-stage regulated studies carry substantially greater regulatory and operational risk compared to failures identified during early-phase development. As a result, the investigation and remediation approach must reflect the level of study impact.
- ResolveMass Laboratories Inc. follows a structured deviation-to-recovery model supported by documented CAPA procedures designed to align with both FDA and EMA regulatory expectations.
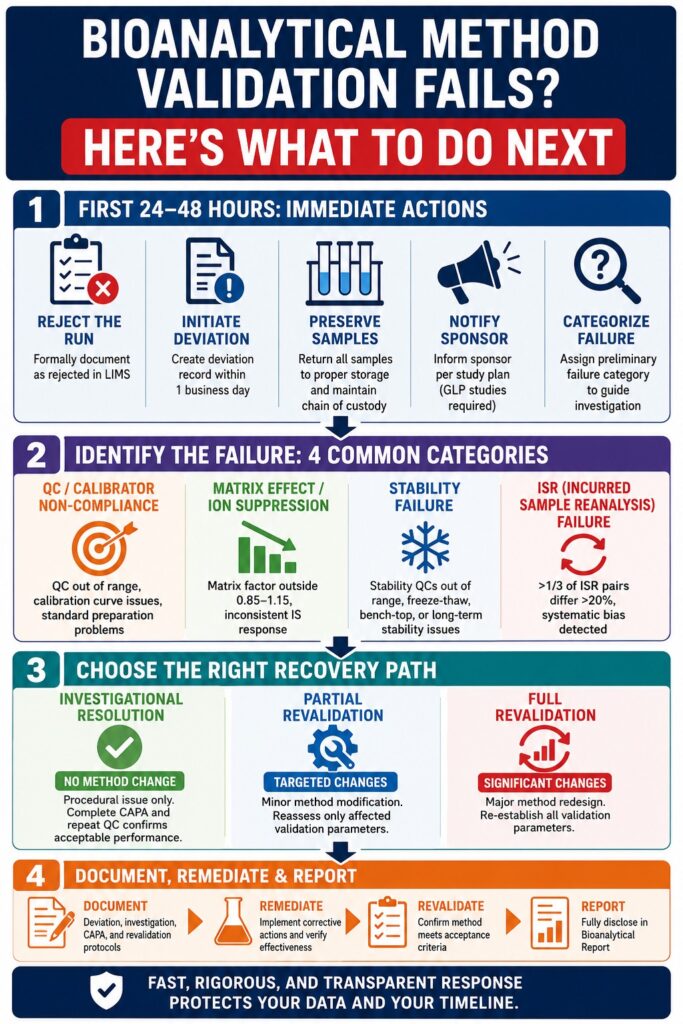
The Moment of Failure: What Must Happen in the First 24–48 Hours
When an analytical run fails to meet acceptance criteria, the first regulatory obligation is to formally reject the run rather than quietly discarding the data. According to the ICH M10 guideline, all failed analytical runs must be reported and discussed in the Bioanalytical Report, with the reasons for failure clearly documented. This initial documentation step forms the basis for every subsequent investigation and recovery action.
Mandatory Immediate Actions
Formal run rejection notation:
The failed analytical run must be formally recorded as rejected within the LIMS (Laboratory Information Management System) rather than being omitted from the dataset. All rejection codes should correspond to pre-defined SOP categories.
Deviation initiation:
A signed deviation record must be initiated within one business day of the failure. This documentation is auditable and cannot be created retrospectively during a regulatory inspection.
Sample integrity preservation:
All study samples associated with the failed run must immediately be returned to appropriate storage conditions. Full chain-of-custody documentation must be maintained throughout the process.
Sponsor notification (for regulated studies):
For GLP-compliant or pivotal studies, sponsors must be informed according to the communication procedures defined in the study plan. This requirement is mandatory.
Preliminary failure categorization:
Based on the parameter that failed, such as calibration curve performance, QC accuracy or precision, selectivity, matrix effect factor, or ISR, the laboratory director must assign a preliminary failure category to guide the scope of the investigation.
Regulatory Caution
Under ICH M10, run rejection criteria must be pre-specified within the SOP or study protocol. Creating a justification retrospectively for excluding a run, without a pre-defined rejection criterion, represents a serious GLP compliance issue. Regulatory inspectors carefully evaluate the distinction between a legitimate run rejection and selective exclusion of unfavorable data.
Mitigate Early Validation Risks: To understand how to proactively structure your testing protocols, review these insights on navigating IND-Enabling Bioanalytical Studies efficiently.
Classifying the Failure: Four Root Cause Categories and Their Diagnostic Pathways
Correctly identifying the type of validation failure is essential because it determines the appropriate recovery strategy. Misclassifying a matrix effect issue as a calibrator preparation problem, for example, can lead to unnecessary troubleshooting efforts and significant delays. The table below outlines the most common bioanalytical method validation failure categories, their diagnostic approaches, and their associated regulatory risks.
| Failure Category | Primary Indicators | Key Diagnostic Steps | Regulatory Risk |
|---|---|---|---|
| QC / Calibrator Non-Compliance | ≥50% of QC samples outside ±15% (±20% at LLOQ); calibration curve r² below acceptance threshold | Re-evaluate reference standard purity, concentration calculations, weighing documentation, diluent preparation, solvent integrity, and pipette calibration logs | Moderate |
| Matrix Effect / Ion Suppression | Normalized matrix factor (NMF) outside 0.85–1.15; inconsistent IS response across matrix lots | Conduct post-column infusion or post-extraction spike experiments; monitor phospholipids; assess sample cleanup efficiency | High |
| Stability Failure | Stability QC values deviate >15% from nominal; freeze-thaw, bench-top, or long-term stability outside specification | Investigate storage temperatures, freeze-thaw cycle counts, container closure integrity, and exposure to light | High |
| ISR (Incurred Sample Reanalysis) Failure | More than one-third of ISR pairs exceed ±20% difference between original and repeat analysis; directional systematic bias | Investigate metabolite back-conversion, in-vitro protein binding changes, sample handling inconsistencies, and instrument drift | High |
The Special Case of ISR Failure
ISR failure requires particular attention because it is one of the leading causes of method invalidation in pivotal studies. A large-scale review of 3,951 regulated bioanalysis studies conducted by the European Bioanalysis Forum found that approximately 1.4% of studies experienced ISR failure. Although relatively uncommon, the consequences were often severe.
Among the failed ISR cases:
- Approximately 70% were resolved after identifying a procedural root cause without requiring changes to the analytical method. Following investigation and CAPA implementation, analysis resumed.
- Around 25% required formal modification and revalidation of the method or analytical procedures before study sample analysis could continue.
- Approximately 5% resulted in complete data rejection, rendering the study data unusable for regulatory submission purposes.
Published CRO investigations have identified several recurring causes of ISR failure, including metabolite back-conversion during sample preparation, particularly with acyl glucuronides and ester prodrugs, differences in protein binding between spiked QC samples and incurred study samples, poor in-injector stability under ambient conditions, and systematic chromatographic integration differences between analysts.
Importantly, FDA guidance clearly states that ISR failures should not be used as a basis for retroactively rejecting original analytical runs. ISR serves as an investigation trigger rather than a direct run rejection criterion.
Avoid Common Pitfalls: Ensure your laboratory protocols align with industry benchmarks by downloading this guide on avoiding Common Bioanalytical Mistakes during routine validation.
Three Recovery Pathways: Investigational, Partial Revalidation, and Full Revalidation
Once the root cause has been identified, the recovery strategy must be formally selected and documented before remediation begins. Under ICH M10, the three available recovery pathways are distinct and cannot be used interchangeably. Choosing an inappropriate pathway may create additional regulatory concerns beyond the original validation failure.
Investigational Resolution
No Method Change Required
This pathway is appropriate when the root cause is procedural in nature, such as a pipetting error, incorrect solvent lot usage, or analyst deviation. Once CAPA activities are completed and repeat QC confirmation demonstrates acceptable performance, analysis may resume without changing the method.
Partial Revalidation
Targeted Method Modification
Partial revalidation applies when a specific aspect of the method has been modified, such as changes to the sample preparation process, chromatographic column, or internal standard concentration. ICH M10 specifies that only the affected validation parameters require reassessment rather than the entire method.
Full Revalidation
Significant Method Redesign
Full revalidation becomes necessary when substantial modifications are introduced, including changes in detection technique, transitions from LLE to SPE extraction procedures, or changes in analyte ionization mode. In these cases, all validation parameters must be re-established.
Execute Smooth Transitions: When modifying an existing assay, it is critical to implement a formal Bioanalytical Method Transfer protocol to avoid total method re-creation.
When Partial Revalidation Is — and Is Not — Defensible
Partial revalidation is often operationally attractive because it reduces time and resource requirements. However, it is also one of the most frequently misapplied recovery strategies. ICH M10 clearly defines the circumstances under which partial validation is appropriate.
Partial revalidation may be justified when:
- Calibration range modifications are minor and do not affect the LLOQ or ULOQ.
- Sample preparation adjustments are isolated and demonstrably non-impactful to method selectivity.
- Matrix changes are limited, such as extending validation to additional matrix lots.
However, partial revalidation is not appropriate when:
- ISR failures reveal systemic selectivity issues.
- Matrix effect failures affect multiple matrix lots.
- Stability failures uncover a fundamental analyte instability mechanism that was not previously characterized.
Best Practice
When determining whether partial or full revalidation is appropriate, the scientific rationale should be thoroughly documented with direct references to the relevant sections of ICH M10 or the FDA 2018 guidance within the revalidation protocol. Regulatory reviewers evaluate whether the selected revalidation scope is proportionate to the nature and risk of the failure rather than simply confirming that some form of revalidation was performed.
Technical Remediation Strategies by Failure Type
Addressing Bioanalytical Method Validation Failure Due to Matrix Effects
Matrix effect failures, especially ion suppression caused by phospholipids in plasma, represent some of the most technically challenging validation issues to resolve. Under ICH M10, the normalized matrix factor (NMF), calculated using the internal standard to compensate for variable suppression, must remain within 0.85–1.15.
When matrix effect criteria are not met, the following corrective actions should be evaluated sequentially:
Enhance Chromatographic Separation
Increasing gradient length or introducing a column wash step can shift phospholipid elution away from the analyte retention window. Because phospholipids are predictably retained on reversed-phase columns, adjusting mobile phase composition can often improve separation efficiency.
Switch or Optimize Sample Preparation
Protein precipitation (PP) carries the highest risk of matrix-related interference. Transitioning to supported liquid extraction (SLE) or solid-phase extraction (SPE) can significantly reduce co-eluting endogenous compounds. However, any modification to the sample preparation procedure requires revalidation of extraction recovery together with matrix effect evaluation.
Optimize the Internal Standard (IS)
A structurally analogous stable-isotope-labeled (SIL) internal standard is considered the gold standard for compensating for variable matrix effects in LC-MS/MS analysis. When a SIL-IS is unavailable, ensuring chromatographic co-elution between the analyte and internal standard becomes essential for proportional compensation of matrix-induced signal changes.
Validate a Dilute-and-Shoot Approach
In certain matrices, validated sample dilution before injection can sufficiently reduce matrix load. When implementing this strategy, dilution integrity must be reassessed during partial revalidation.
Secure Method Integrity: If you are working with complex human matrices or cerebrospinal fluid, discover specialized Tissue and CSF Bioanalytical Services engineered to overcome tough matrix challenges.
Addressing Stability Failures
Stability failures are especially serious because they may raise retrospective concerns regarding the integrity of all study samples analyzed under the affected conditions. For example, if freeze-thaw stability fails after two or more cycles and study samples have already undergone those cycles, the bioanalytical report must discuss whether the resulting stability gap compromises pharmacokinetic conclusions.
Short-Term (Bench-Top) Failure
Investigations should focus on temperature excursions, excessive sample processing times, and unintended light exposure. SOPs should be revised to shorten allowable bench-top exposure windows, followed by re-demonstration of stability within the revised timeframe.
Freeze-Thaw Failure
The investigation should assess the analyte’s chemical stability during repeated freeze-thaw cycles. High-risk compounds include acyl glucuronides, nitro compounds, and certain biologics. Laboratories may need to reduce the maximum allowable freeze-thaw cycles permitted within sample handling SOPs.
Long-Term Storage Failure
Long-term stability failure represents one of the most operationally severe events. If study samples have exceeded their validated storage stability window, all associated PK data may be compromised. A complete stability reinvestigation should begin immediately, and the sponsor must be informed without delay.
Addressing Calibration and QC Acceptance Failures
If more than 50% of QC samples within a run fall outside ±15% of nominal concentration, or ±20% at the LLOQ, the analytical run is considered invalid. Repeated occurrences across multiple validation runs indicate a systematic issue rather than random analytical variability.
The investigation should include the following:
Reference Standard Integrity Verification
Review the certificate of purity, storage conditions, reconstitution records, and weighing documentation. Degradation of a reference standard in solution is among the most common causes of recurring QC failure.
Instrument Performance Review
Evaluate MRM transition area responses, source cleanliness, and detector response drift over the relevant analysis period. Ion source contamination in mass spectrometers accumulates during routine operation and progressively reduces sensitivity.
Analyst Variability Investigation
Compare QC performance across analysts if multiple personnel participated in the study. Systematic pipetting bias is a recognized contributor to precision failure and may not be immediately apparent from instrument-level assessments.
Regulatory Documentation and Reporting During a Bioanalytical Method Validation Failure Event
Technical remediation without appropriate documentation does not constitute a compliant recovery process. The following documentation activities must proceed in parallel with laboratory investigations.
| Document | When Required | Content Requirements |
| Deviation Record | Within 1 business day of failure | Description of the failure, affected runs, preliminary categorization, investigation plan, responsible personnel |
| Root Cause Investigation Report | Within 5–10 business days | Detailed root cause analysis, supporting evidence, impact assessment, corrective actions |
| CAPA (Corrective and Preventive Action) | Before resuming analysis | Short-term correction and long-term prevention measures, implementation timeline, verification criteria |
| Partial/Full Revalidation Protocol | Before initiating revalidation studies | Scope justification, parameters to reassess, acceptance criteria, applicable guidance references |
| Updated Bioanalytical Report Section | In final study report | Full disclosure of the failure, investigation findings, corrective actions, and impact on data integrity |
Ensure Compliance Standards: For a deeper understanding of compliance thresholds across agencies, read about the core EMA vs FDA Bioanalytical Method Validation Differences to align your documentation perfectly.
Critical Regulatory Note
Both ICH M10 and FDA guidance require that any deviation from pre-defined procedures be disclosed within the Bioanalytical Report. This requirement is mandatory. Failure to report validation issues represents a serious data integrity violation and is one of the most significant findings during FDA inspections of bioanalytical laboratories.
Any chromatogram reintegration, repeat injection, or study sample reanalysis must be documented together with the scientific rationale for performing the action.
Maintain Uncompromising Standards: Learn how to build defensible compliance records by focusing heavily on Data Integrity in Bioanalytical Studies.
Cross-Validation Implications After Failure
When a method failure occurs during an ongoing study or during transfer between laboratories, cross-validation may be necessary before regulated sample analysis can resume. ICH M10 emphasizes statistically driven cross-validation approaches such as Bland-Altman analysis or Deming regression rather than relying solely on simple pass/fail comparisons.
If the method was modified during recovery, data generated under the original method must be evaluated for comparability with data generated under the corrected method. This consideration becomes particularly important in NDA and ANDA submissions where pharmacokinetic data spans multiple study phases.
Clinical Stage Matters: How Regulatory Consequences Scale With Development Phase
The consequences of a bioanalytical method validation failure vary considerably depending on the stage of drug development. The table below summarizes the expected impact and corresponding urgency of response.
| Development Phase | Validation Standard | Failure Consequence | Recovery Priority |
| Discovery / Preclinical Screening | Fit-for-purpose | Minimal regulatory impact; redesign is common | Low urgency |
| IND-Enabling / Phase I | Full validation (FDA 2018 / ICH M10) | Potential clinical program delays; dosing may be paused if sample integrity is affected | High urgency |
| Pivotal PK / BE Studies | Full validation (GLP) | Possible study invalidation; regulatory notification may be required; NDA timelines impacted | Critical |
| Post-Approval / Comparative BE | Full validation (GLP) | Increased risk of ANDA rejection; possible study repetition with 12–24 month delay | Critical |
Navigate Late-Phase Requirements: Discover tailored strategies for large clinical trials by viewing our dedicated Bioanalytical CRO Services for Phase II & Phase III to secure your market-entry path.
How ResolveMass Laboratories Inc. Manages Validation Failure Events
At ResolveMass Laboratories Inc., validation failures are managed through a structured deviation-to-recovery framework that treats every failure as a quality system event rather than merely a laboratory setback. The framework immediately activates three parallel workstreams once a failure is confirmed: technical investigation, regulatory documentation, and sponsor communication.
LC-MS/MS platforms are operated using validated SOPs covering run rejection criteria, ISR investigation triggers, and CAPA lifecycle management, all aligned with ICH M10 and FDA 2018 guidance. When ISR failure occurs, the standard investigation protocol includes systematic evaluation of metabolite back-conversion, protein binding variability between spiked and incurred samples, in-injector stability under study conditions, and inter-analyst precision variability.
CAPA closure is not approved until the investigation report has been scientifically justified and independently reviewed by qualified personnel outside the original analytical team.
For sponsors facing a critical bioanalytical method validation failure during a pivotal study, ResolveMass Laboratories Inc. also provides regulatory strategy support, including assessments regarding whether the impact on existing study data requires formal FDA disclosure and how revalidation scope should be documented to withstand regulatory review.
Establish a Resilient Alliance: Learn how a collaborative, quality-first Bioanalytical CRO Partnership can help your organization seamlessly manage and prevent validation roadblocks.
Conclusion: Bioanalytical Method Validation Failure Is Manageable With the Right Framework
A bioanalytical method validation failure does not automatically terminate a development program. The final outcome depends on the speed, rigor, and quality of the response: how quickly the failure is documented, how thoroughly the root cause is identified, how appropriately the recovery pathway is defined, and how transparently the event is reported within the bioanalytical documentation package.
These factors are not secondary administrative concerns. They are among the primary criteria regulators evaluate when reviewing failure events associated with NDA, ANDA, or IND submissions.
Whether the failure involves ISR non-reproducibility, matrix effect issues that escaped initial screening, calibrator integrity problems, or stability window exceedances, the scientific and regulatory recovery framework is clearly established within ICH M10. The challenge lies in execution, where laboratory experience, quality systems, and regulatory alignment determine whether a project recovers efficiently or experiences delays extending 12 to 24 months.
For organizations navigating a bioanalytical method validation failure within a regulated study environment, the most important next step is to treat the event as a formal quality system issue requiring structured investigation rather than as a technical inconvenience that can simply be corrected and restarted.
Frequently Asked Questions: Bioanalytical Method Validation Failure
Yes, a study can usually continue even if a single validation run fails. The failed run must first be formally rejected according to pre-established SOP criteria, and a documented root cause investigation must be initiated immediately. If the issue is traced to a procedural or isolated operational error that can be corrected, and subsequent QC performance meets acceptance criteria, sample analysis may proceed. However, if the investigation identifies a broader method-related problem affecting reliability or reproducibility, the study analysis must pause until appropriate revalidation activities are completed.
According to ICH M10 and FDA 2018 bioanalytical guidance, an analytical run becomes invalid if more than 50% of QC samples fall outside the acceptable accuracy limits of ±15%, or ±20% at the LLOQ. In addition, acceptable QC performance must include at least one passing concentration level from both the lower and upper portions of the calibration range. Failure to satisfy these conditions requires formal run rejection and documented investigation. These criteria are designed to ensure consistency, reliability, and overall analytical integrity throughout the study.
Partial revalidation is performed when only a limited part of the analytical method has been modified, such as changes to sample preparation, chromatographic conditions, or internal standard concentration. In these situations, only the affected validation parameters require reassessment. Full revalidation, in contrast, involves repeating the complete validation process from the beginning because major aspects of the method have changed. This typically applies when there are modifications involving the analytical technique, biological matrix, analyte characteristics, or detection platform that may impact overall method performance.
When a stability failure is identified after study samples have already been analyzed under the affected conditions, concerns arise regarding the reliability of the associated pharmacokinetic data. The laboratory must conduct a formal scientific impact assessment to determine whether sample degradation could have influenced analytical results. The findings and conclusions must be documented within the bioanalytical report, and sponsors in regulated studies must be informed promptly. Depending on the severity of the issue and remaining sample availability, reanalysis of affected samples may become necessary.
ISR failure is not technically identical to a method validation failure, but it is still considered highly significant. Incurred Sample Reanalysis is performed after validation to confirm that the method produces reproducible results when analyzing actual study samples rather than spiked QC materials. A failed ISR indicates that the method may not be consistently reliable under real study conditions, which raises concerns about its suitability for the intended application. FDA guidance emphasizes that ISR failures should trigger a structured investigation rather than automatic rejection of previously accepted analytical runs.
There is no universal requirement for immediate real-time FDA notification following every validation failure event. However, the failure, associated investigation, corrective actions, and final resolution must be fully disclosed within the bioanalytical documentation submitted as part of the NDA, ANDA, or BLA package. If the failure significantly impacts data integrity or compromises critical study outcomes, sponsors may need to evaluate whether additional regulatory communication, protocol amendments, or clinical hold considerations are necessary. Regulatory affairs teams should be involved as early as possible during pivotal study failures.
Recovery timelines for full method revalidation vary depending on assay complexity, analyte properties, and matrix requirements. For a standard small molecule LC-MS/MS assay, complete redevelopment, optimization, and revalidation activities commonly require approximately 6 to 12 weeks. This timeframe includes reassessment of selectivity, accuracy, precision, matrix effect, stability, calibration range, and sensitivity parameters. Ligand binding assays (LBAs) and large molecule bioanalytical methods may require additional time due to reagent qualification, antibody characterization, and assay variability considerations.
Published investigations from CROs and findings from the European Bioanalysis Forum consistently identify metabolite back-conversion as one of the leading causes of ISR failure in LC-MS/MS assays. This issue is especially common with acyl glucuronides and ester-containing compounds that may chemically convert during sample handling or preparation. Another major contributor involves differences in protein binding behavior between freshly prepared QC samples and actual incurred study samples. Instrument response drift and variability between original and repeat analytical runs are also recognized as important contributing factors.
Reference:
- U.S. Food and Drug Administration. (2018, May). Bioanalytical method validation guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry
- U.S. Food and Drug Administration. (2022, November). M10 bioanalytical method validation and study sample analysis: Guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m10-bioanalytical-method-validation-and-study-sample-analysis
- European Medicines Agency. (2022). ICH guideline M10 on bioanalytical method validation and study sample analysis – Step 5 (EMA/CHMP/ICH/172948/2019). https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf

