
Introduction:
A robust CMC Strategy for Complex Generic Injectables is one of the most important factors influencing successful ANDA approval. Unlike conventional oral generics, complex injectable products—including long-acting injectables (LAIs), liposomes, microspheres, emulsions, suspensions, peptide formulations, polymer-based depots, and nanoparticle drug products—require extensive analytical characterization and process understanding to demonstrate pharmaceutical equivalence.
Regulatory agencies such as the FDA increasingly expect sponsors to establish deep scientific knowledge of the reference listed drug (RLD), identify critical formulation attributes, and develop manufacturing processes capable of consistently reproducing product quality. This requires analytical expertise that extends well beyond routine quality control testing.
An experienced analytical CRO/CDMO such as ResolveMass Laboratories Inc. supports pharmaceutical companies by providing advanced characterization, reverse engineering, formulation support, extractables and leachables testing, bioanalytical services, and regulatory-ready analytical documentation throughout the CMC lifecycle.
Summary:
- A strong CMC strategy for complex generic injectables must address formulation equivalence, particle/microsphere characterization, sterility assurance, and a defensible regulatory data package from day one — not as an afterthought before filing.
- Complex generics (long-acting injectables, liposomes, PLGA microspheres, nanosuspensions) face higher FDA/Health Canada scrutiny than small-molecule generics because they require proof of in vitro and in vivo equivalence to the Reference Listed Drug (RLD), not just identical active ingredient content.
- A capable CDMO partner should bring orthogonal analytical methods, robust reference standard qualification, stability program design, and CMC documentation that survives regulatory review the first time.
- Common failure points include inadequate particle size/morphology comparison, incomplete impurity and leachables profiling, weak justification of critical quality attributes (CQAs), and stability protocols that don’t reflect the intended commercial container/closure system.
- Timeline and cost planning should account for the fact that complex generic injectable CMC packages typically require 12–24 months of comparative characterization and stability work before a filing is submission-ready.
- ResolveMass Laboratories supports sponsors across formulation characterization, method development, and regulatory-ready CMC data generation for complex generic injectable programs.
1: What Makes CMC Strategy Different for Complex Generic Injectables?
A CMC strategy for complex generic injectables differs from standard generics because regulators require demonstrated sameness across formulation, structure, and release performance — not just chemical identity. For products like depot suspensions, liposomal injectables, and PLGA-based microspheres, the FDA’s product-specific guidances (PSGs) typically demand comparative physicochemical characterization, in vitro release testing (IVRT), and often clinical pharmacokinetic bridging.
This means CMC planning starts earlier and runs deeper than it would for an oral generic tablet. Formulation scientists and analytical teams need to work from the same data set, because a gap in particle size distribution data or an unqualified reference standard can stall a filing by months. This is one of the main reasons sponsors increasingly look at outsourcing generic drug development to a partner with dedicated complex-generics infrastructure rather than building this capability in-house.
Sponsors evaluating a generic drug development CDMO for a complex injectable program should look specifically for experience with long-acting formulations. A CDMO for long-acting injectable formulation development brings a fundamentally different skill set than one focused on conventional solution or suspension injectables, since depot and sustained-release products introduce structural variables that standard formulation review doesn’t typically address. In Canada specifically, sponsors working with a CDMO for generic drug development in Canada also need to account for Health Canada’s own comparative data expectations alongside FDA requirements when the program targets both markets.
Complex generic injectables generally fall into a handful of well-defined categories, each with distinct CMC expectations:
| Product Type | Examples | Key CMC Focus Areas |
|---|---|---|
| PLGA/PLA microsphere depots | Naltrexone, aripiprazole, goserelin, leuprolide | Particle size, polymer molecular weight, drug distribution, burst release |
| Liposomal injectables | Doxorubicin liposomal, amphotericin B liposomal | Lamellarity, encapsulation efficiency, vesicle size distribution |
| Nanosuspensions/nanocrystals | Injectable antipsychotic suspensions | Particle size distribution, crystal form, redispersibility |
| Iron colloid complexes | Iron sucrose, ferric carboxymaltose | Molecular weight distribution, colloid stability, carbohydrate shell structure |
| Long-acting implants | Subdermal drug-eluting implants | Polymer degradation kinetics, drug release mechanism |
Each category carries its own FDA product-specific guidance (PSG), and Health Canada applies comparable expectations through its own generic drug submission requirements. A CMC strategy that treats all complex injectables the same way — rather than tailoring the characterization plan to the specific product class — is one of the more common early planning mistakes.
Peptide-based injectables deserve a specific mention here. Products like GLP-1 receptor agonists and leuprolide depots sit at the intersection of peptide chemistry and complex formulation science, and sponsors developing these programs typically need a partner offering dedicated peptide CDMO services alongside standard formulation characterization. For GLP-1 programs specifically, GLP-1 peptide analytical characterization requires methods capable of resolving closely related sequence variants and degradation products, while leuprolide depot generics benefit from a CRO for leuprolide depot development that understands both the peptide and the PLGA delivery matrix. Nanosuspension-based injectables raise a related but distinct challenge, since poorly soluble APIs formulated as nanocrystals require particle engineering expertise; sponsors working on these should look for CDMO experience specific to generic drug development for poorly soluble APIs.
Why Do Complex Injectables Draw More Regulatory Scrutiny?
Complex injectables draw more scrutiny because their in vivo performance is driven by physical and structural attributes that are difficult to replicate exactly, not just by active pharmaceutical ingredient (API) potency. A generic sponsor can match the API 1:1 and still fail bioequivalence if the microsphere size distribution, polymer degradation rate, or liposome lamellarity differs meaningfully from the RLD.
Regulators such as the FDA’s Office of Generic Drugs (OGD) evaluate these products under a “weight of evidence” approach, combining:
- Physicochemical characterization (particle size, morphology, polymorphism)
- In vitro release/dissolution profiles under biorelevant conditions
- Formulation composition (Q1/Q2) and, where required, Q3 structural comparison
- Clinical PK data demonstrating bioequivalence
The Q1/Q2/Q3 framework is central to understanding scrutiny levels. Q1 (qualitative sameness) means identical inactive ingredients; Q2 (quantitative sameness) means matching concentrations within acceptable limits; Q3 (structural/physicochemical sameness) applies specifically to complex products where the arrangement of the formulation matters as much as its composition. For PLGA microspheres, for example, two formulations can be Q1/Q2 equivalent — same polymer, same drug loading — and still fail Q3 comparison if the internal drug distribution or polymer degradation profile differs. This is precisely why complex generics can’t rely on the compendial dissolution-testing shortcuts available to oral solid dosage generics.
Sponsors should also expect that OGD review teams weigh the internal consistency of the data package as much as any single result. A characterization dataset that shows tight agreement across orthogonal methods — for instance, particle size confirmed independently by laser diffraction and image analysis — carries more evidentiary weight than a single-method result, even if both individually meet a specification.
Given how much this scrutiny varies by product class and jurisdiction, sponsors often benefit from dedicated regulatory support for generic drugs in the US and Canada early in program planning rather than treating regulatory strategy as a downstream filing activity. This is particularly true for peptide-containing complex injectables, where peptide drug regulatory requirements can differ meaningfully from small-molecule expectations around impurity qualification and sequence-related variants.
2: What Should a CDMO Deliver for Complex Generic Injectable CMC Packages?
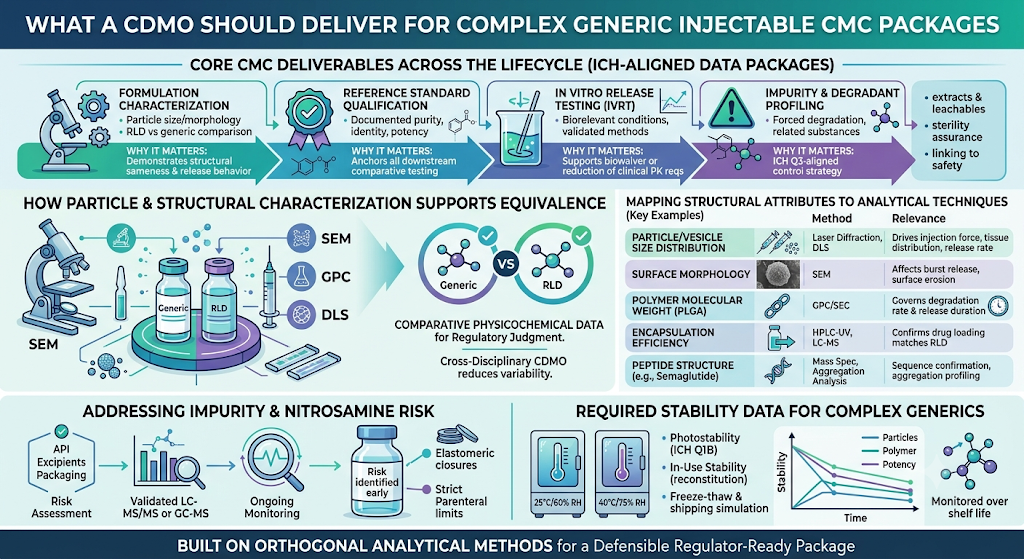
A CDMO supporting complex generic injectable development should deliver a defensible, regulator-ready CMC data package built on orthogonal analytical methods, not a single point-of-truth technique. Relying on one method to characterize a critical quality attribute is a common reason review cycles stall, because agencies expect confirmatory data from complementary techniques. This is why robust analytical development for generic drugs — built around method validation and cross-technique confirmation — sits at the center of any credible CMC strategy for this product class.
The table below outlines core deliverables a CDMO should provide across the CMC lifecycle for a complex generic injectable program.
| CMC Area | Typical Deliverable | Why It Matters |
|---|---|---|
| Formulation characterization | Particle size/morphology comparison (RLD vs. generic) | Demonstrates structural sameness driving release behavior |
| Reference standard qualification | Certified reference standards with documented purity, identity, potency | Anchors all downstream comparative testing |
| In vitro release testing (IVRT) | Method development and validation under biorelevant conditions | Supports waiver or reduction of clinical PK requirements |
| Impurity and degradant profiling | Forced degradation, related substances, nitrosamine risk assessment | Required for ICH Q3-aligned impurity control strategy |
| Extractables & leachables | Container closure and delivery device compatibility studies | Confirms product safety across shelf life |
| Stability program | ICH-conditioned long-term and accelerated stability data | Supports proposed shelf life and storage claims |
| Sterility assurance | Endotoxin, sterility, and container closure integrity testing | Non-negotiable for parenteral products |
How Does Particle and Structural Characterization Support Equivalence?
Particle and structural characterization supports equivalence by generating the comparative physicochemical data regulators use to judge whether a generic behaves like the RLD. For PLGA microsphere depots, this typically includes particle size distribution (laser diffraction), morphology (SEM), porosity, and polymer molecular weight/degradation profile. For liposomal products, lamellarity, encapsulation efficiency, and vesicle size distribution are central comparative attributes.
A CDMO with cross-disciplinary analytical capability — mass spectrometry, chromatography, and imaging under one roof — can generate these datasets with consistent sample handling, which reduces variability that reviewers might otherwise flag as inconclusive.
The table below maps common structural attributes to the analytical techniques typically used to characterize them, along with why each attribute matters for equivalence claims.
| Structural Attribute | Typical Method(s) | Relevance to Equivalence |
|---|---|---|
| Particle/vesicle size distribution | Laser diffraction, dynamic light scattering | Drives injection force, tissue distribution, and release rate |
| Surface morphology | Scanning electron microscopy (SEM) | Affects burst release and surface erosion behavior |
| Polymer molecular weight | Gel permeation chromatography (GPC/SEC) | Governs degradation rate and total release duration |
| Encapsulation efficiency | HPLC-UV, LC-MS | Confirms drug loading matches RLD within acceptable range |
| Crystal form/polymorphism | X-ray powder diffraction (XRPD), DSC | Impacts solubility, dissolution, and long-term stability |
| Internal drug distribution | Confocal microscopy, micro-CT | Indicates whether drug is surface-associated or core-encapsulated |
For PLGA-based products specifically, polymer characterization deserves particular attention. The lactide:glycolide ratio, end-group chemistry (acid-capped vs. ester-capped), and molecular weight distribution all influence hydrolytic degradation rate, which in turn drives the release profile. A generic that matches the RLD’s initial particle size but uses a polymer with a different end-group chemistry can still diverge significantly in release kinetics over the dosing interval — a gap that only shows up in extended in vitro release studies run over the full intended duration of action (which, for some depot products, can mean multi-month testing protocols). Sponsors evaluating leuprolide depot CDMO selection specifically should weigh this polymer-behavior expertise heavily, since it’s one of the more technically demanding depot categories to characterize comparatively.
Where the API itself is a peptide, structural characterization extends further to include sequence confirmation, related-substance profiling, and peptide degradation product characterization, since degradation pathways for peptides differ substantially from small-molecule APIs. Dedicated peptide analytical characterization services — covering mass spectrometry-based sequencing, oxidation/deamidation profiling, and aggregation analysis — are typically required alongside the formulation-level structural work described above. ResolveMass’s own semaglutide characterization case study illustrates how this type of layered characterization approach applies in practice for a GLP-1 peptide program.
How Should Impurity and Nitrosamine Risk Be Addressed in the CMC Package?
Impurity and nitrosamine risk should be addressed through a documented risk assessment aligned to ICH M7 and current FDA/Health Canada nitrosamine guidance, supported by validated, sufficiently sensitive analytical methods. For injectables specifically, this includes assessing leachables from elastomeric components, secondary amines in formulation excipients, and process-related nitrosamine precursors.
A defensible approach generally includes:
- A documented nitrosamine risk assessment covering API synthesis route, excipients, and packaging
- Validated LC-MS/MS or GC-MS methods with sensitivity appropriate to acceptable intake limits
- Ongoing monitoring built into the control strategy, not a one-time study
For injectables, nitrosamine risk assessment also needs to account for interaction between the formulation and the primary packaging — rubber stoppers, elastomeric closures, and certain coating materials can introduce secondary amines or nitrite sources that react over shelf life. This is distinct from the API-focused nitrosamine assessments common in oral solid dosage generics, and it’s a step sponsors sometimes underscope early in development. A CDMO offering dedicated nitrosamine testing for CDMO programs should be able to demonstrate method sensitivity appropriate to parenteral acceptable intake limits, which are often more stringent than oral product thresholds.
What Stability Data Does a Complex Generic Injectable Filing Require?
A complex generic injectable filing requires ICH-conditioned stability data collected on the actual intended commercial packaging configuration, covering both long-term and accelerated conditions, plus in-use stability if the product is multi-dose or requires reconstitution. Stability programs for these products typically need to demonstrate that structural attributes — not just chemical potency — remain within specification over the proposed shelf life.
Key elements of a defensible stability program include:
- Long-term (typically 25°C/60% RH) and accelerated (40°C/75% RH) conditions per ICH Q1A(R2), with intermediate conditions added where the product shows accelerated sensitivity
- Photostability testing per ICH Q1B where the formulation or primary packaging offers limited light protection
- Structural attribute monitoring across the stability timeline (e.g., particle size drift, polymer molecular weight decline, aggregation) — not potency and impurities alone
- In-use stability studies simulating actual clinical handling, including reconstitution, dilution, and multi-dose withdrawal where applicable
- Freeze-thaw and shipping simulation studies for products with cold-chain distribution requirements
3: What Are the Most Common CMC Gaps That Delay Complex Generic Injectable Filings?
The most common CMC gaps that delay filings are insufficient comparative characterization data, unqualified or poorly documented reference standards, and stability data that doesn’t reflect real-world container/closure combinations. Each of these triggers information requests (deficiency letters) that can add six months or more to a review timeline.
Sponsors can reduce this risk by involving their CDMO early in:
- Defining critical quality attributes (CQAs) against the RLD before formulation lock
- Building a method validation package that anticipates ICH Q2(R2) expectations
- Structuring stability protocols around the intended commercial packaging configuration
- Documenting a clear, audit-ready CAPA process for any out-of-specification results
- Establishing a reference standard qualification protocol before comparative testing begins, since every downstream equivalence claim depends on it
The table below summarizes the gap, its typical root cause, and the downstream regulatory consequence sponsors most often encounter.
| Common Gap | Root Cause | Downstream Consequence |
|---|---|---|
| Incomplete comparative characterization | Reliance on a single analytical method for a CQA | Information request asking for orthogonal confirmation |
| Unqualified reference standard | RLD material sourced without documented purity/identity verification | Entire comparative dataset called into question |
| Non-representative stability packaging | Stability studies run on lab-scale containers, not commercial configuration | Requirement to repeat stability program on correct packaging |
| Underscoped nitrosamine assessment | Risk assessment focused only on API, excluding packaging interactions | Additional risk assessment and testing requested post-submission |
| Insufficient IVRT duration | Release testing stopped before full intended duration of action | Data judged insufficient to support release profile match |
Each of these gaps is avoidable with early planning, but they share a common thread: they surface late, usually during agency review, at a point where remediation costs months rather than weeks. Building the characterization plan around the specific PSG (or, in its absence, a scientifically justified comparative approach) before formulation lock is the single highest-leverage step a sponsor can take.
4: Why Does CDMO Selection Matter for CMC Strategy Execution?
CDMO selection matters because CMC strategy is only as strong as the analytical and regulatory documentation behind it — a partner without deep characterization capability and Health Canada/FDA-aligned quality systems introduces risk into the filing timeline. Sponsors should evaluate a CDMO’s experience with complex parenteral dosage forms specifically, not general small-molecule generics, since the analytical demands are materially different.
Key evaluation criteria include:
- Demonstrated experience with PLGA, liposomal, or long-acting injectable characterization
- Breadth of orthogonal analytical techniques (mass spectrometry, chromatography, imaging, particle sizing)
- Track record supporting ANDA/regulatory submissions with CMC data that has withstood agency review
- Quality systems that can generate GMP-aligned, audit-ready documentation
- Capacity to run extended-duration in vitro release studies without becoming a scheduling bottleneck
- Willingness to engage in early formulation-phase consultation rather than only executing predefined test plans
Sponsors evaluating CDMOs should also ask pointed questions during the selection process: How many complex parenteral generic programs has the lab supported through to filing? What orthogonal methods does it use to confirm particle size and morphology data? How does it structure reference standard qualification, and can it produce a full chain-of-custody record? A CDMO that answers these specifically — rather than in general capability terms — is more likely to deliver documentation that withstands review.
5: How ResolveMass Laboratories Supports Complex Generic Injectable CMC Programs
ResolveMass Laboratories works with generic drug sponsors to build the analytical backbone of a CMC strategy for complex generic injectables, spanning particle and structural characterization, impurity and nitrosamine testing, reference standard qualification, and stability program support. Our team applies orthogonal mass spectrometry and chromatographic methods so that comparative data holds up under regulatory scrutiny, rather than relying on a single technique that leaves gaps for reviewers to question.
Our analytical scope for complex generic injectable programs typically includes:
- Mass spectrometry-based structural and impurity characterization
- Particle size and morphology analysis using complementary techniques
- Polymer characterization (molecular weight distribution, end-group chemistry) for PLGA-based products
- Nitrosamine and extractables/leachables testing aligned to current FDA and Health Canada expectations
- Reference standard qualification with full documentation supporting comparative claims
- ICH-conditioned stability program design and execution
For sponsors navigating PLGA microsphere depots, liposomal injectables, or other complex parenteral generics, involving an experienced analytical CDMO early in program design is one of the most effective ways to reduce filing risk and review cycles.
Conclusion:
A successful CMC Strategy for Complex Generic Injectables requires far more than manufacturing capability. It demands a comprehensive understanding of formulation science, analytical characterization, process development, regulatory expectations, and lifecycle quality management.
As regulatory standards continue to evolve, pharmaceutical companies benefit from partnering with analytical experts capable of generating robust, scientifically defensible CMC data. An experienced CDMO with advanced characterization capabilities can significantly improve development efficiency, reduce regulatory risk, and strengthen the overall quality of an ANDA submission.
For companies developing long-acting injectables, polymer-based depots, liposomes, microspheres, suspensions, or other complex injectable products, investing in a well-planned CMC Strategy for Complex Generic Injectables is essential for achieving successful commercialization.
Frequently Asked Questions:
A robust CMC strategy ensures that complex generic injectables consistently match the quality and performance of the reference listed drug (RLD). These products often contain sophisticated drug delivery systems such as liposomes, microspheres, or polymer-based depots, where even small manufacturing variations can affect clinical performance. A comprehensive CMC strategy helps identify critical quality attributes (CQAs), optimize manufacturing processes, establish validated analytical methods, and generate regulatory-ready documentation. This minimizes development risks, supports regulatory approval, and reduces costly delays during the review process.
Reverse engineering provides detailed insights into the formulation and manufacturing characteristics of the reference listed drug. Through advanced analytical characterization, scientists can identify the API properties, excipient composition, polymer grades, particle morphology, residual solvents, degradation products, and manufacturing signatures. This information guides formulation development, process optimization, and analytical method selection. A well-executed reverse engineering strategy reduces development uncertainty and helps sponsors establish pharmaceutical equivalence more efficiently.
Quality by Design (QbD) is a systematic development approach that builds product quality into the manufacturing process rather than relying solely on end-product testing. It involves identifying Critical Quality Attributes (CQAs), Critical Process Parameters (CPPs), and Critical Material Attributes (CMAs), followed by risk assessments and design of experiments (DoE). Applying QbD principles enables manufacturers to optimize formulations, improve process robustness, reduce variability, and establish a scientifically justified control strategy, ultimately supporting smoother regulatory approvals.
A Contract Development and Manufacturing Organization (CDMO) provides scientific, analytical, and manufacturing expertise throughout the product development lifecycle. An experienced CDMO supports reverse engineering, formulation optimization, analytical method development and validation, process scale-up, stability studies, extractables and leachables testing, and regulatory documentation. By offering integrated CMC services, a CDMO helps pharmaceutical companies reduce development timelines, mitigate technical risks, and prepare high-quality submissions for regulatory agencies.
Both the FDA and EMA expect sponsors to provide comprehensive scientific evidence demonstrating product quality, manufacturing consistency, and pharmaceutical equivalence. CMC submissions should include detailed formulation information, manufacturing process descriptions, validated analytical methods, impurity characterization, stability studies, process validation data, container closure evaluations, and risk assessments. Regulatory authorities also encourage Quality by Design (QbD) principles and robust lifecycle management strategies to ensure continued product quality after approval.
Stability studies demonstrate that an injectable product maintains its quality, potency, purity, and safety throughout its intended shelf life. These studies typically include long-term, accelerated, stress, photostability, freeze-thaw, and in-use evaluations under ICH-recommended conditions. Critical parameters such as assay, degradation products, pH, sterility, particle size, appearance, and drug release profile are monitored over time. Stability data are essential for establishing expiration dates, storage conditions, and regulatory approval.
When selecting a CDMO, pharmaceutical companies should evaluate technical expertise, analytical capabilities, regulatory experience, quality systems, instrumentation, and experience with similar complex injectable products. The CDMO should be capable of providing advanced characterization, formulation support, validated analytical methods, stability testing, extractables and leachables studies, and CTD-ready documentation. Strong communication, project management, and a proven regulatory track record are also important considerations for successful product development.
Reference
- Thakur K, Kaur SD, Kak D, Kapoor DN. Quality by Design in parenteral drug development: Addressing formulation challenges and industrial insights. Drug Development and Industrial Pharmacy. 2026 Feb 6(just-accepted):1-23.https://www.tandfonline.com/doi/abs/10.1080/03639045.2026.2628937
- Gutierrez L, Cauchon NS, Christian TR, Giffin MJ, Abernathy MJ. The confluence of innovation in therapeutics and regulation: recent CMC considerations. Journal of Pharmaceutical Sciences. 2020 Dec 1;109(12):3524-34.https://www.sciencedirect.com/science/article/pii/S0022354920305530
- Adler M, Design DD, Goli VA, Allmendinger A, Mahler HC, Officer CE. Transitioning from Vial to Subcutaneous Injection Devices for Biological Drug Products. Published online. 2024 May(9):2024.https://www.ondrugdelivery.com/wp-content/uploads/2024/05/160_2024_May_Injectables_ten23_health.pdf
- Chen D, Zhang X, Xu S, Liu D. Current CMC challenges in oligonucleotide API development: from IND to NDA. Medicinal Chemistry Research. 2026 Apr 25:1-5.https://link.springer.com/article/10.1007/s00044-026-03569-8

