
Executing a rigorous program for Extractables & Leachables (E&L) Testing for transdermal drug delivery systems is a core regulatory requirement because migrating chemical impurities from multilayer patch components and secondary packaging can directly penetrate the skin barrier. This creates the possibility of systemic toxicological exposure as well as localized dermal sensitization risks. By identifying and quantifying these impurities under simulated clinical conditions, manufacturers demonstrate product safety, quality assurance, and regulatory compliance throughout the entire product lifecycle.
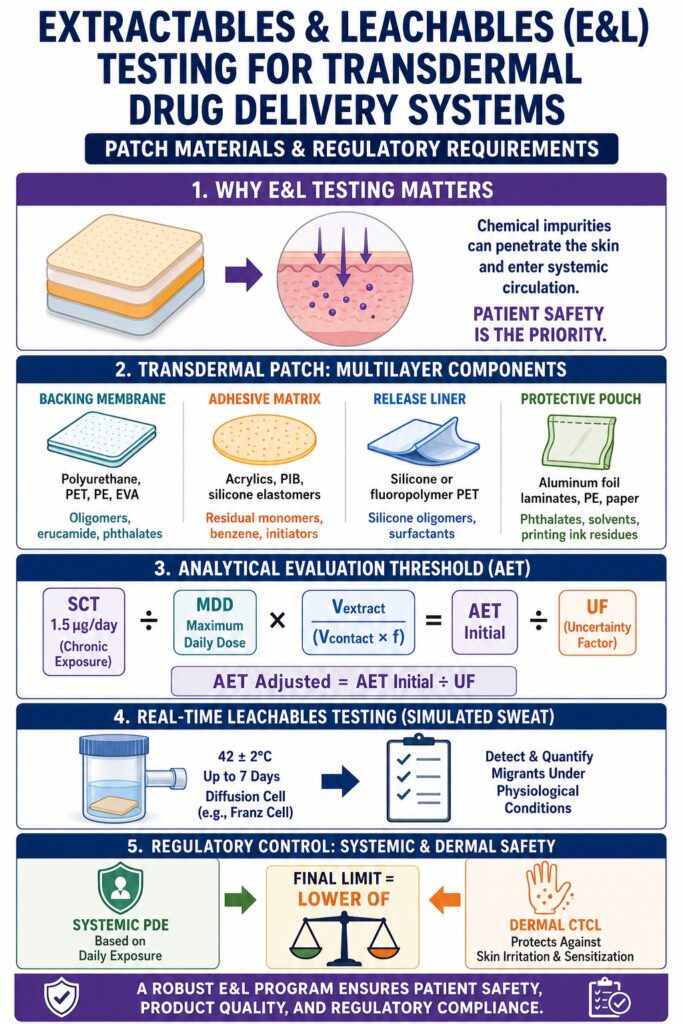
Transdermal delivery systems (TDS) are categorized by global regulatory authorities, including the FDA and EMA, as high-risk drug-device combination products. Unlike oral dosage forms, where the gastrointestinal tract reduces exposure to many non-volatile contaminants, or topical products that remain on the skin surface, transdermal systems deliver active pharmaceutical ingredients (APIs) through the stratum corneum and into systemic circulation. As a result, any chemical migrant that partitions from patch materials into the adhesive layer may be delivered directly to the patient, increasing exposure risk.
To ensure patient safety, regulatory expectations for transdermal systems have evolved significantly. The United States Pharmacopeia (USP) has introduced chapter <1664.4>, which specifically addresses topical and transdermal drug products and complements USP <1663> (extractables) and USP <1664> (leachables). Internationally, the International Council for Harmonisation (ICH) has proposed guideline Q3E, which provides a harmonized, risk-based framework for E&L evaluation across regulatory regions. For developers submitting New Drug Applications (NDAs) or Abbreviated New Drug Applications (ANDAs), a complete chemical characterization profile is essential to avoid regulatory delays, including Complete Response Letters (CRLs).
To avoid unexpected regulatory delays, explore our comprehensive guide on the root causes of failed extractables and leachables E&L studies.
Share via:
Article Summary:
- Extractables & Leachables (E&L) testing is a critical regulatory requirement for transdermal drug delivery systems because chemical migrants from patch materials or packaging can penetrate skin and enter systemic circulation, posing safety risks.
- Regulatory agencies like the FDA and EMA classify transdermal patches as high-risk combination products, requiring strict compliance with guidelines such as USP <1663>, USP <1664>, USP <1664.4>, and the emerging ICH Q3E framework.
- Transdermal patches consist of multiple polymer layers (backing films, adhesives, release liners, and packaging), each of which can release chemical impurities such as monomers, plasticizers, solvents, and catalyst residues under storage or use conditions.
- Pressure-sensitive adhesives (PSAs) are especially prone to migration due to their flexible structure, which accelerates diffusion of low-molecular-weight compounds, especially under the influence of permeation enhancers and storage conditions.
- Analytical evaluation relies on defined thresholds like AET (Analytical Evaluation Threshold), which converts toxicological limits into measurable analytical concentrations to ensure sensitive detection of potentially harmful substances.
- Controlled extractables and leachables studies use aggressive solvents, elevated temperatures, and advanced techniques (GC-MS, LC-MS, ICP-MS) to build a comprehensive profile of possible migrants and simulate worst-case exposure scenarios.
- Regulatory safety assessment combines systemic exposure limits (PDE) and local dermal toxicity limits (CTCL/DST), ensuring that both whole-body toxicity and skin sensitization risks are controlled before product approval.
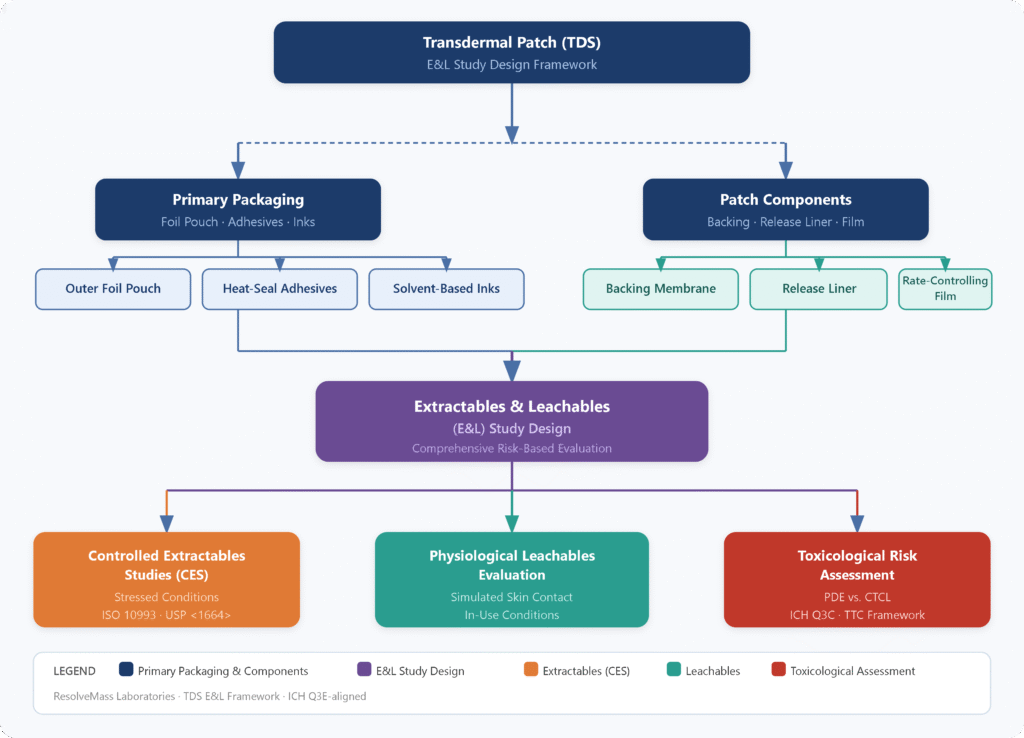
What are the Primary Materials and Polymer Dynamic Concerns in Transdermal Patches?
Transdermal patches are constructed from complex multilayer polymer systems that include backing films, pressure-sensitive adhesive (PSA) matrices, rate-controlling membranes, release liners, and protective pouch packaging. The high free volume and low glass transition temperatures (Tg) of PSAs promote thermodynamic migration of low-molecular-weight compounds, residual monomers, and processing aids into the drug matrix.
Understanding material composition is essential for identifying contamination sources. Each component contributes potential leachables:
Backing Films: Typically made of polyethylene terephthalate (PET), ethylene-vinyl acetate (EVA), polyurethane, or polyethylene. These layers prevent outward drug loss but may contain degradation products, catalyst residues, and slip agents such as erucamide.
Pressure-Sensitive Adhesives (PSAs): Composed of acrylics, polyisobutylenes (PIBs), or silicone-based polymers. These systems may contain residual monomers such as 2-ethylhexyl acrylate, methyl methacrylate, acrylic acid, cross-linking agents, and initiator residues such as benzoyl peroxide and its degradation product benzene.
Release Liners: Silicone- or fluoropolymer-coated polyester films designed to protect adhesive surfaces prior to application. These can release silicone oligomers, fluorinated surfactants, and unreacted curing agents.
Protective Pouches: Multilayer foil systems using polyurethane tie layers and printed outer films. Residual solvents, plasticizers such as phthalates, and photoinitiators from printing inks may migrate into the adhesive layer through reverse migration or absorption.
Because PSAs exist in an amorphous, highly mobile state, contaminant diffusion is significantly faster than in rigid polymers. The inclusion of permeation enhancers such as fatty acids, surfactants, and terpenes further plasticizes the system, lowering Tg and increasing free volume. This accelerates migration from secondary packaging into the adhesive during storage.
Discover how modern material selection can mitigate migration risks by reading about low leachables packaging materials.
Component Layer Overview
| Component | Polymer Materials Used | Representative Migrants (E&L) | Primary Toxicological & Quality Risks |
|---|---|---|---|
| Backing Membrane | Polyurethane, PET, polyethylene, EVA | Oligomers, erucamide, phthalates | Discoloration, systemic toxicity |
| Adhesive Matrix | Acrylics, PIB, silicone elastomers | Residual monomers, benzene, initiators | Skin irritation, sensitization, carcinogenic risk |
| Release Liner | Silicone or fluoropolymer PET | Silicone oligomers, surfactants | Adhesion failure, dermal irritation |
| Protective Pouch | Aluminum foil laminates, polyethylene, paper | Phthalates, solvents, PAHs | Endocrine disruption, mutagenicity |
How is the Analytical Evaluation Threshold (AET) Calculated for Transdermal Patches?
The Analytical Evaluation Threshold (AET) is derived by converting the systemic Safety Concern Threshold (SCT), typically 1.5 micrograms per day for chronic exposure, into a measurable analytical concentration based on maximum daily dose (MDD) and extraction conditions. This ensures that analytical methods are sensitive enough to detect potentially harmful migrants.
For a deeper breakdown of threshold determinations, review our expert analysis on calculating the AET for extractables and leachables studies.
The calculation links toxicological thresholds with dosing and analytical parameters.
Mathematical Equations for AET Derivation
Under USP <1664.4> and PQRI principles, the SCT for chronic transdermal exposure is 1.5 micrograms per day. For shorter exposure scenarios, values such as 5.0 micrograms per day or 50 micrograms per day may apply depending on risk justification.
The initial AET is calculated as:
AET initial = (SCT / MDD) × (Vextract / (Vcontact × f))
Where:
- SCT represents Safety Concern Threshold in micrograms per day
- MDD represents Maximum Daily Dose
- Vextract represents solvent volume used in extraction
- Vcontact represents contact surface area or material mass
- f represents accumulation factor for multiple components
For dose-based systems:
AET estimated = (SCT / Doses per day) × (Doses CCS / Weight CCS)
Because GC-MS and LC-MS methods vary in detector response, an uncertainty factor (UF) is applied:
AET adjusted = AET initial / UF
In GC-EI-MS, UF is often set to 2 due to stable response behavior. In LC-ESI-MS, UF is derived from relative response factor variability:
UF = 1 / (1 – RSD) or UF = Mean / (1 – (t × Std))
If variability increases significantly, UF may approach infinity, indicating the method requires optimization or segmentation into targeted assays.
Learn how to optimize your testing array by reading our technical review on GC-MS vs LC-MS in extractables and leachables testing.
How are Controlled Extractables Studies (CES) Conducted for Transdermal Components?
Controlled extractables studies involve isolating each patch layer and subjecting it to aggressive extraction conditions using solvents and elevated temperatures to generate a worst-case chemical profile. This forms a reference library for future leachables comparison.
Solvent selection typically includes:
- Water (polar phase)
- 50% ethanol or isopropanol (intermediate phase)
- Hexane (non-polar phase)
- Nitric acid solutions for elemental analysis
For detailed strategies on selecting appropriate media, refer to our article on solvents for extractables studies.
Casting solvents such as ethyl acetate or toluene are also used to simulate manufacturing conditions.
Extraction is performed at 40°C to 55°C for 24 to 72 hours using sonication, reflux, or Soxhlet methods. Surface area to solvent volume ratios of 3 cm²/mL to 6 cm²/mL are commonly applied.
After extraction, samples undergo multi-platform analysis:
- Headspace GC-MS for volatile compounds
- GC-MS for semi-volatiles
- LC-MS/MS for non-volatiles
- ICP-MS for elemental impurities
This ensures detection of additives, stabilizers, plasticizers, and UV absorbers at trace levels.
Explore how trace metals are quantified with high precision in our guide on ICP-MS in extractables and leachables testing.
How are Real-Time and Simulated Dermal Leachables Studies Performed?
Real-time and simulated studies evaluate chemical migration under physiological conditions using a sweat-based matrix maintained at 42 ± 2°C. This replicates skin temperature under stress conditions.
Unlike static container systems, transdermal patches are influenced by sweat, occlusion, and blood flow, all of which alter diffusion behavior.
The simulated sweat matrix typically contains sodium chloride, lactic acid, urea, and organic modifiers such as ethanol or isopropanol to replicate sebum interactions.
Two main experimental approaches are used:
Whole-Patch Immersion: Entire patch is submerged after removing the liner. This may overestimate exposure.
Single-Sided Extraction: Uses diffusion cells such as Franz cells or ASTM F739 systems to isolate the adhesive surface and better simulate real use.
USP <1664.4> defines surface area to volume ratios:
- Thin patches (<0.5 mm): 6 cm²/mL
- Thick patches (>0.5 mm): 3 cm²/mL
- Hydrogel patches: 0.5–1.0 cm²/mL
Extraction is performed at 42 ± 2°C for the full wear duration, which may extend up to seven days. Recovery validation targets 50% to 80% to ensure analytical reliability.
Learn how to build long-term evaluation protocols with our insights on leachables monitoring during stability studies.
How do Regulatory Bodies Control Elemental Impurities and Localized Sensitization Risks?
Regulatory frameworks manage risks by comparing systemic Permitted Daily Exposure (PDE) values with localized Cutaneous Toxicity Concentration Limits (CTCL), applying the stricter requirement.
Under USP <1664.4> and ICH Q3D(R2), transdermal products require both systemic and dermal safety evaluations.
Systemic exposure is calculated as:
C systemic = Transdermal PDE / MDD
The final limit is the lower value between systemic PDE-based limits and CTCL.
For example, cobalt has a PDE of 50 micrograms per day and a CTCL of 35 micrograms per gram. If MDD is 1.0 g/day, the systemic limit is 50 micrograms per gram, but CTCL governs at 35 micrograms per gram. If MDD increases to 2.0 g/day, systemic limit becomes 25 micrograms per gram and becomes the controlling factor.
Palladium PDE is also used as a surrogate for related platinum group metals, ensuring broader catalyst control.
For organic compounds, ICH Q3E introduces the Dermal Sensitization Threshold (DST) of 1 microgram per cm² per day (approximately 500 ppm). Exposure below DST is generally considered low risk, while exceedances require full toxicological evaluation including route-to-route extrapolation using F6 factors.
Understand the formal evaluation paths for organic and inorganic impurities by reviewing our resource on the toxicological qualification of leachables.
Element & Toxicity Class
| Element | Transdermal PDE (μg/day) | CTCL (μg/g) | Governing Limit for 1.0 g/day MDD | Core Source in Patch Materials |
|---|---|---|---|---|
| Cadmium (Class 1) | 20 | None established | 20 μg/g | Plastic stabilizers, pigments |
| Lead (Class 1) | 50 | None established | 50 μg/g | Colorants, recycled plastics |
| Arsenic (Class 1) | 30 | None established | 30 μg/g | Mineral fillers |
| Mercury (Class 1) | 30 | None established | 30 μg/g | Catalyst residues |
| Cobalt (Class 2A) | 50 | 35 [cite: 4] | 35 μg/g (CTCL-driven) | Pigments, catalysts |
| Vanadium (Class 2A) | 200 | 35 [cite: 4] | 35 μg/g (CTCL-driven) | Polymer catalysts |
| Nickel (Class 2A) | 100 | 35 [cite: 4] | 35 μg/g (CTCL-driven) | Metal coatings, catalysts |
Conclusion: Achieving Regulatory Compliance in Extractables & Leachables (E&L) Testing for Transdermal Delivery Systems
Establishing a scientifically robust Extractables & Leachables (E&L) program for transdermal drug delivery systems is essential for aligning advanced patch technologies with regulatory safety expectations. Because these systems operate under continuous skin contact and dynamic physiological conditions, generic testing approaches are not sufficient.
Compliance with USP <1664.4> and execution of physiologically relevant simulation studies remain critical for regulatory acceptance and timely approvals.
Ensure your regulatory submission data meets strict trace compliance standards by reviewing data integrity in extractables and leachables testing.
ResolveMass Laboratories Inc. develops tailored chemical characterization strategies designed to meet FDA and ICH expectations. By integrating advanced chromatographic techniques with toxicological risk assessment, the organization supports developers in managing complex transdermal polymer systems.
For instance, developers can compare these parameters with our cross-industry frameworks for E&L testing for pre-filled syringes
.
For customized study design and regulatory consultation on container-closure compatibility for transdermal systems, contact ResolveMass Laboratories Inc. through its official channels.
Frequently Asked Questions (FAQs) on Extractables & Leachables (E&L) Testing for Transdermal Drug Delivery
In transdermal systems, AET is calculated based on solid-state parameters such as patch surface area or dry mass of the adhesive matrix. This is different from parenteral products, where calculations are tied to liquid dose volumes expressed in μg/mL. For patches, the unadjusted AET (AET_initial) is typically expressed in μg/g of material. The equation incorporates factors like maximum daily dose and extraction volume to reflect solid polymer behavior. This approach ensures analytical methods are properly aligned with extractables originating from non-liquid dosage forms.
The adhesive casting solvent is included because traces of it may remain trapped within the cured polymer network. These residues can swell the adhesive matrix and significantly influence the mobility of extractable compounds. When the same solvent is used in controlled extraction studies, it better simulates the internal chemical environment of the patch. This helps release compounds that would otherwise remain inaccessible under standard conditions. As a result, the study more accurately reflects real-world leachable behavior during storage and use.
Reservoir-type patches contain a liquid or gel compartment, which increases the risk of migration from membranes, seals, and backing materials into the drug reservoir. This design can also introduce risks like leakage or dose dumping if structural integrity is compromised. In contrast, matrix-type patches embed the drug within a solid adhesive system, reducing leakage risk. However, matrix systems tend to absorb external contaminants such as plasticizers or ink residues from packaging components. Both systems present different but significant E&L challenges.
USP <1664.4> requires evaluation of both systemic exposure limits (PDE) and localized skin toxicity limits (CTCL), with the stricter value governing final specifications. For cobalt and vanadium, systemic PDE values are higher, but both share a lower CTCL threshold of 35 μg/g. When applied to a 1.0 g/day patch, the CTCL becomes the controlling limit. This ensures that dermal sensitization risks are controlled even when systemic exposure remains within acceptable ranges. The framework prioritizes patient safety at the site of application.
Matrix interference in transdermal systems is commonly addressed using sample clean-up techniques such as solid-phase extraction (SPE) and liquid-liquid extraction (LLE). These methods help separate analytes from adhesive polymers, surfactants, and other interfering substances. After clean-up, high-sensitivity techniques like LC-MS/MS or GC-MS are applied for detection. This workflow reduces ion suppression and improves signal clarity. As a result, even trace-level leachables can be reliably identified and quantified.
Single-sided extraction is preferred because it replicates real patient exposure more accurately. Only the adhesive side of the patch is exposed to the simulated biological medium, while the backing and non-contact layers are isolated. This prevents overestimation of leachables that would never interact with the skin. Whole-patch immersion can artificially inflate detected compounds from non-relevant surfaces. Therefore, single-sided methods provide more clinically meaningful results.
ICH Q3E introduces adjustment factors such as F6 to convert oral toxicity data into dermal exposure estimates. These factors account for differences in absorption between ingestion and skin contact. For example, low oral bioavailability leads to higher uncertainty factors during extrapolation. This ensures that dermal Permitted Daily Exposure (PDE) values remain protective even when starting from oral toxicology data. The approach improves consistency in cross-route safety assessments.
A three-tier approach evaluates the adhesive at different stages: raw polymer, intermediate laminate form, and final finished patch. This stepwise method helps isolate the origin of extractables at each stage of production. It distinguishes impurities from raw materials, curing reactions, or packaging migration. By separating these sources, regulators and manufacturers can better understand contamination pathways. This improves both product control and regulatory transparency.
Reference:
- Kuzmič, S., Zlobec, T., & Trdan Lušin, T. (2025). Extractables and leachables in pharmaceutical products: Potential adverse effects and toxicological risk assessment. Pharmaceuticals.
https://pmc.ncbi.nlm.nih.gov/articles/PMC12846058/ - Sharma, A., Kumar, R., & Patel, S. (2025). [Title not clearly displayed on landing view of PMC record]. PubMed Central (PMC). https://pmc.ncbi.nlm.nih.gov/articles/PMC12967098/
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2025). ICH Q3E: Guideline for extractables and leachables (Step 2 draft presentation, 26 August 2025). https://database.ich.org/sites/default/files/ICH_Q3E_Step2_Presentation_2025_0826.pdf
- European Medicines Agency. (2025). Draft ICH Q3E guideline for extractables and leachables. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-q3e-guideline-extractables-leachables_en.pdf
- United States Pharmacopeia. (n.d.). Extractables and leachables. https://www.usp.org/impurities/extractables-and-leachables

