
Introduction:
Implementation of Nitrosamine Testing for Injectable Drug Products is a critical safety and regulatory requirement because parenteral drug products bypass the body’s natural protective physiological barriers. As a result, even trace levels of mutagenic impurities can pose serious risks to patient health. This highly specialized analytical testing process is used to identify and quantify extremely low-level carcinogenic impurities, including nitrosamines and drug substance-related impurities, in sterile liquid, suspension, and lyophilized formulations.
By combining advanced high-resolution mass spectrometry with comprehensive risk-based evaluations, pharmaceutical developers can establish strong control strategies that meet the expectations of global health authorities. ResolveMass Laboratories Inc. provides advanced analytical testing services that help developers manage the complex thermodynamic and chemical challenges associated with parenteral formulation development.
Need a partner for your analytical needs? Nitrosamine method development and validation services.
Article Summary:
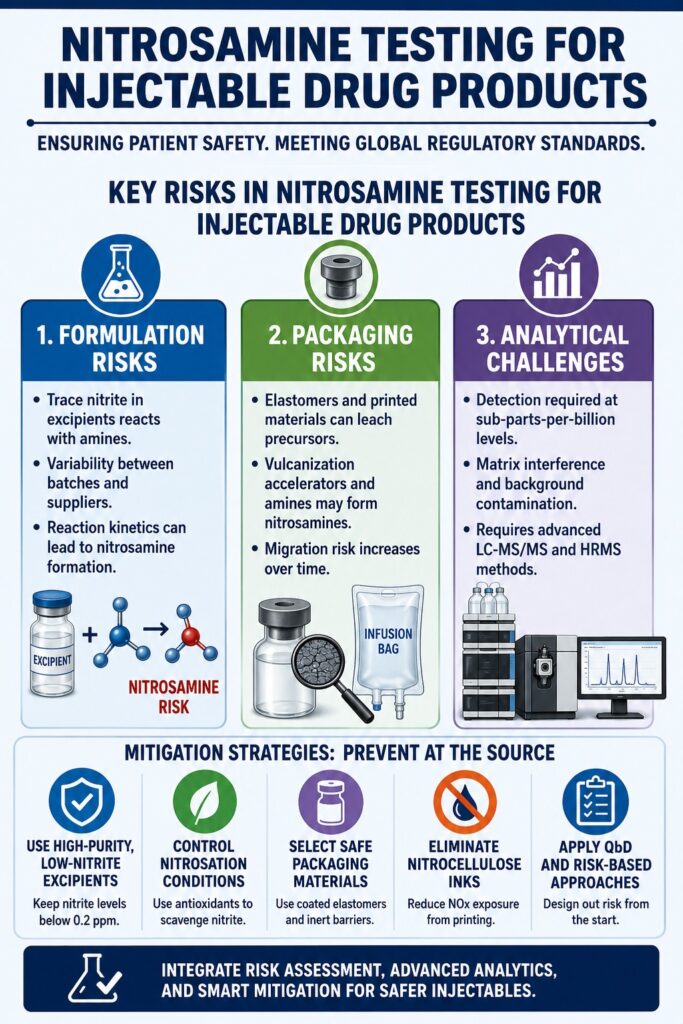
- Nitrosamine testing is essential for injectable drug products because these formulations enter the bloodstream directly, making even trace amounts of carcinogenic impurities a potential safety concern. Regulatory agencies require highly sensitive testing to protect patients.
- Global authorities such as the FDA, EMA, and Health Canada mandate a structured risk management process that includes risk assessment, confirmatory testing, and mitigation to ensure nitrosamine levels remain within established acceptable intake limits.
- Formulation components can significantly influence nitrosamine formation. Trace nitrites, residual amines, excipient variability, and manufacturing conditions may trigger unwanted reactions, making careful raw material selection and supplier control essential.
- Packaging materials are another important source of nitrosamine risk. Rubber closures, infusion bags, tubing, and printed packaging components may release precursor compounds that can migrate into injectable formulations during storage.
- Advanced analytical technologies such as LC-MS/MS and high-resolution mass spectrometry (HRMS) enable reliable detection of nitrosamines at ultra-trace levels, while specialized workflows help overcome matrix interference and improve analytical accuracy.
- Regulatory compliance depends on robust method validation following USP and ICH Q2(R2) guidelines, demonstrating specificity, accuracy, precision, sensitivity, and reproducibility suitable for regulatory submissions.
- Preventive manufacturing strategies provide the most effective long-term solution. Using low-nitrite excipients, optimizing formulation design, selecting inert packaging materials, and applying Quality-by-Design (QbD) principles help minimize nitrosamine formation and support global regulatory compliance.
Regulatory Requirements for Nitrosamine Testing for Injectable Drug Products
Global regulatory authorities require a three-step risk management approach consisting of risk evaluation, confirmatory testing, and mitigation to ensure that nitrosamine impurities remain below defined Acceptable Intake (AI) limits. These requirements are outlined in the FDA’s September 2024 final guidance, the EMA Article 5(3) opinion, and Health Canada directives, all aimed at ensuring parenteral products do not present carcinogenic risks to patients.
Unsure if your product requires evaluation? Do all drugs need nitrosamine risk assessment?
Because nitrosamines are classified as Class 1 mutagenic carcinogens, their toxicity at extremely low concentrations demands strict control at acceptable daily intake levels in the nanogram range. These limits are far below the standard Threshold of Toxicological Concern (TTC) of 1.5 micrograms/day.
Under current FDA and EMA frameworks, manufacturers must assess whether the API, excipients, manufacturing processes, or storage conditions can lead to nitrosamine formation. When compound-specific carcinogenicity data are not available, regulators recommend using the predicted Carcinogenic Potency Categorization Approach (CPCA) to establish safe exposure limits based on molecular structure. For products involving multiple doses or long-term administration, these daily limits are adjusted according to the maximum daily dose (MDD) of the drug product to calculate allowable concentrations in parts per million (ppm) or parts per billion (ppb).
Globally Aligned Acceptable Intake (AI) Limits for High-Risk Nitrosamine Impurities
| Nitrosamine Impurity | CAS Number | Maximum Intake – FDA (ng/day) | Maximum Intake – EMA (ng/day) | Potency Category / Toxicity Source |
|---|---|---|---|---|
| N-Nitrosodimethylamine (NDMA) | 62-75-9 | 96 | 96 | Category 1 / Highly potent |
| N-Nitrosodiethylamine (NDEA) | 55-18-5 | 26.5 | 26.5 | Category 1 / Highly potent |
| N-Nitrosoethylisopropylamine (NEIPA) | 16339-04-1 | 400 | 400 | Process-related synthetic risk |
| N-Nitrosodiisopropylamine (NDIPA) | 601-77-4 | 26.5 | 1500 | Manufacturing/solvent impurity |
| N-Nitroso-N-methyl-4-aminobutyric acid (NMBA) | 61445-55-4 | 1500 | 1500 | Commonly found in sartan drugs |
| N-Nitrosodibutylamine (NDBA) | 924-16-3 | 26.5 | 26.5 | Packaging overwrap/elastomer leachable |
Formulation Challenges: Excipient Nitrite Variability and Reaction Kinetics
A major formulation challenge involves controlling trace nitrite and amine impurities present in pharmacopeial-grade excipients, as these act as limiting reagents in nitrosamine formation. Even when excipients meet compendial purity standards, residual nitrite levels below 1 ppm can react with secondary or tertiary amines in APIs, producing regulated nitrosamines during manufacturing or storage.
Explore the chemistry: Nitrosamine formation pathways in API synthesis.
Managing this risk requires a detailed understanding of how excipient-specific chemical properties influence nitrosation kinetics.
Batch-to-batch and supplier-to-supplier variability in excipients represents a significant scientific challenge. For instance, microcrystalline cellulose (MCC) from different suppliers may contain nitrite levels ranging from 0.15 ppm to 0.85 ppm. When combined with an amine-containing API, these differences can significantly alter nitrosamine formation rates during accelerated stability testing.
Polymeric excipients such as polyvinylpyrrolidone (PVP) and hydroxypropyl methylcellulose (HPMC) may also contain residual secondary or tertiary amine precursors from synthesis solvents or catalysts. Under acidic conditions or thermal stress, these residues can initiate internal nitrosation reactions even without direct API involvement.
Trace Nitrite Levels and Nitrosamine Risk Potential Across Parenteral Excipients
| Excipient Type | Average Nitrite Range (ppm) | Primary Nitrosation Pathway / Mechanism | Nitrosamine Formation Risk |
|---|---|---|---|
| Polyethylene Glycol (PEG) | 0.20 – 1.50 | Oxidative degradation produces aldehydes and peroxides | High |
| Polyvinylpyrrolidone (PVP) | 0.50 – 2.00 | Residual amine catalysts from polymerization | High |
| Lactose Monohydrate | 0.10 – 0.50 | Water-based nitrite contamination during processing | Moderate |
| Microcrystalline Cellulose (MCC) | 0.05 – 0.90 | Trace nitrites from raw materials and bleaching | Low |
| Polysorbate 80 | 0.10 – 0.80 | Autoxidation and residual amine-related reactions | Moderate |
Packaging Risks in Nitrosamine Testing for Injectable Drug Products
Container closure systems can act as indirect sources of nitrosamine contamination because elastomers and printed flexible foils may leach precursor compounds into drug formulations. Evaluating these pathways requires extensive extractables and leachables (E&L) studies under real-time and accelerated conditions to ensure system integrity.
Since injectable products remain in prolonged contact with packaging materials, the risk of chemical migration increases over time.
Elastomeric stoppers and plungers present a particularly high risk due to vulcanization accelerators such as thiurams and dithiocarbamates used in rubber manufacturing. These compounds can degrade into secondary amines, which then react with nitrite residues within the elastomer to form extractable nitrosamines.
Clarify the risks: Nitrosamine impurity vs. Nitrosamine leachable difference.
This interaction is influenced by the physical state of the product. Lyophilized vial formulations often show higher levels of rubber-stopper leachables upon reconstitution compared to liquid formulations. This is due to changes in partitioning behavior and physical-chemical stresses at the interface between the rubber stopper and lyophilized cake during freezing and long-term storage.
For large-volume parenterals in flexible bags, regulatory agencies have identified printed overwraps and pouches as important sources of nitrosamines such as NDBA. Solvent-based printing inks containing dialkylamines may react with nitrogen oxides (NOx) generated from nitrocellulose binders during high-temperature sealing processes. The resulting volatile nitrosamines can migrate through polymer layers into sterile infusion fluids.
Packaging Component Vulnerabilities and Chemical Migration Pathways
| Packaging Component | Potential Leachable / Precursor | Chemical Pathway to Nitrosamine Formation | Relative Migration Risk |
|---|---|---|---|
| Polypropylene Infusion Bags | Amine-based stabilizers / slip agents | Slow migration of secondary amines into liquid matrix | High |
| Rubber Stoppers / Plungers | Vulcanization accelerators (thiurams) | Reaction of amines with nitrite residues within elastomer | Moderate to High |
| Infusion Bag Tubing | Nitrosatable plasticizers | Contact-based extraction under continuous flow | High |
| Printed Aluminum Foil Seals | Adhesive residues, low molecular weight amines | Reaction with gas-phase NOx from nitrocellulose layers | Moderate |
Analytical Solutions: Advanced LC-MS/MS and HRMS Protocols
Quantitative detection of nitrosamines at sub-parts-per-billion levels is performed using liquid chromatography-tandem mass spectrometry (LC-MS/MS) and high-resolution mass spectrometry (HRMS). These techniques provide the sensitivity and selectivity required to distinguish trace impurities from complex formulation matrices. ResolveMass Laboratories Inc. applies specialized workflows designed specifically for sterile parenteral products.
In liquid injectables, matrix effects from excipients, APIs, and high-salt buffers can interfere with detection. For example, in 0.9% sodium chloride intravenous solutions, high salt concentrations may suppress ionization and contaminate mass spectrometry sources. To address this, solvent diversion systems are used to route salt-rich fractions to waste before analytes reach the detector.
Need expert oversight for your testing? Outsourcing nitrosamine testing to a CRO.
Trace nitrosamine analysis is also vulnerable to background contamination from solvents, tubing, and mobile phases. To minimize this, a delay column is installed between the pump and injector, allowing background impurities to separate temporally from analytes of interest.
Additionally, optimized “soft transmission” settings in triple quadrupole instruments reduce ion fragmentation in the source, improving detection of unstable low-molecular-weight nitrosamines such as NDMA and NMBA.
USP Method Validation and Compliance Criteria
Method validation under USP General Chapter and ICH Q2(R2) ensures that analytical procedures are reliable, reproducible, and suitable for regulatory submission. Validation must demonstrate specificity, accuracy, precision, and appropriate limits of quantitation for each matrix.
Since acceptable daily intake levels are in the nanogram range, even small analytical variations can significantly impact regulatory outcomes.
Ensure your submission is ready: Nitrosamine risk assessment for ANDA submission.
USP General Chapter defines multiple validated approaches. Procedure 1 uses LC-HRMS, while Procedure 3 uses LC-MS/MS to target key nitrosamines in sartan drug products. Manufacturers are increasingly adopting a lifecycle approach to validation, maintaining Analytical Procedure Lifecycle Files (APLFs) that document method development, performance trends, and continuous improvements.
USP and ICH Q2(R2) Method Validation Acceptance Criteria
| Validation Parameter | USP Reference Criteria | Analytical Goal / Execution |
|---|---|---|
| Specificity | No chromatographic or isobaric interference at retention times | Resolve nitrosamines from APIs and degradation products |
| Linearity | R² ≥ 0.99 over range of 0.5× to 2.0× specification limit | Ensure proportional response across at least 5 levels |
| Accuracy | Recovery of 70% – 130% (80% – 120% preferred at specification) | Spike-and-recovery in representative matrices |
| Repeatability | RSD ≤ 15% (RSD ≤ 20% at LOQ) | Minimum n=6 replicate injections |
| Limit of Detection (LOD) | Signal-to-noise ratio (S/N) ≥ 3:1 | Detect down to 0.03 ng/mL |
| Limit of Quantitation (LOQ) | S/N ≥ 10:1, RSD ≤ 20% | Reliable quantification at trace levels |
Formulation and Manufacturing Mitigation Strategies
Effective mitigation requires changes in manufacturing design and material selection to eliminate nitrosamine precursors rather than attempting removal after formation. Parenteral developers are encouraged to apply Quality-by-Design (QbD) principles to prevent impurity formation at the source.
At the formulation stage, the use of high-purity, low-nitrite excipients is essential. Incoming raw materials should be controlled to maintain nitrite levels below 0.2 ppm. Where elimination is not feasible, antioxidants such as ascorbic acid or alpha-tocopherol may be used to scavenge nitrite species. These compounds reduce nitrous acid (HNO2) to nitric oxide (NO), preventing reaction with susceptible amines.
From a packaging perspective, elastomers with advanced barrier coatings are preferred. Ethylene tetrafluoroethylene (ETFE) coatings, such as PremiumCoat®, create an inert barrier that reduces leaching of vulcanization accelerators and organic residues into drug products.
Need a strategy for your product? Nitrosamine reformulation strategy.
For flexible container systems, eliminating nitrocellulose-based printing inks reduces exposure to NOx species generated during sealing processes, thereby lowering the risk of nitrosamine formation after packaging.
Conclusion: Achieving Analytical Defensibility in Nitrosamine Testing
Regulatory compliance in parenteral drug development requires the integration of ultra-trace analytical methods with comprehensive risk assessment of both excipients and packaging systems. Due to the severe systemic risks associated with injectable exposure, regulatory authorities expect complete, highly sensitive, and scientifically justified datasets.
Incomplete risk assessments or insufficiently sensitive analytical methods are not acceptable for regulatory approval.
Engaging an experienced contract research organization helps ensure that Nitrosamine Testing for Injectable Drug Products meets global standards for accuracy, reproducibility, and data integrity. ResolveMass Laboratories Inc. provides customized method development, full ICH-compliant validation, and regulatory documentation support to strengthen product safety submissions.
Meet your deadlines with confidence: Nitrosamine testing timeline.
For specialized support with nitrosamine risk assessments, custom analytical method development, or batch release testing, developers can contact the ResolveMass Laboratories Inc. technical team through its official consultation channels.
Sponsors requiring assistance with flexible container overwrap evaluations or USP compliance audits can submit sample requests via the ResolveMass technical consultation portal.
For analytical validation frameworks or custom reference standard coordination, direct support is available through ResolveMass Laboratories Inc. assistance services – Contact us.
Frequently Asked Questions
Injectable drug products enter the bloodstream directly, bypassing the gastrointestinal tract and first-pass liver metabolism. This direct systemic exposure removes key biological protection mechanisms that normally reduce toxicity. As a result, even extremely small traces of nitrosamines can pose a higher carcinogenic risk in parenteral formulations. Regulatory expectations are therefore much stricter, with significantly lower acceptable detection and exposure limits compared to oral dosage forms.
Packaging components, especially elastomeric parts, may contain residual vulcanizing agents such as thiurams. These compounds can degrade over time and generate secondary amines within the container closure system. Once formed, these amines can migrate into the drug solution and react with trace nitrites or nitrogen oxides (NOx), leading to nitrosamine formation. This indirect pathway makes packaging a critical factor in overall product safety.
NDBA contamination in flexible infusion systems is mainly associated with secondary packaging materials, including printed overwraps and protective pouches. During manufacturing, high temperature and pressure conditions can break down nitrocellulose-based ink binders, producing nitrogen oxides. These reactive species then interact with dialkylamines present in adhesives or inks, forming volatile NDBA. The compound can subsequently migrate through polymer layers into the final infusion product.
Regulatory agencies recommend highly sensitive chromatographic techniques coupled with advanced mass spectrometry. Commonly used platforms include LC-MS/MS, GC-MS/MS, and high-resolution mass spectrometry systems such as Orbitrap-based instruments. These technologies are capable of detecting nitrosamines at ultra-trace levels in the parts-per-billion range. They also provide the selectivity needed to reduce interference from complex parenteral matrices.
USP Procedure 1 is based on liquid chromatography coupled with high-resolution mass spectrometry (LC-HRMS) and is designed for broad multi-analyte detection. In contrast, Procedure 3 uses LC-MS/MS and focuses specifically on six targeted nitrosamines commonly found in sartan drug substances. Procedure 1 offers broader screening capability, while Procedure 3 provides targeted, highly sensitive quantification for defined impurities.
Managing nitrite variability begins with strict control of raw material quality and supplier qualification programs. Manufacturers should set tight incoming specifications, typically limiting nitrite content to below 0.2 ppm. Consistent supplier sourcing helps reduce batch-to-batch variation in excipients. In addition, formulation scientists may include nitrite-scavenging antioxidants to further reduce the risk of nitrosamine formation during manufacturing and storage.
Freeze-drying introduces extreme processing conditions, including freezing, vacuum exposure, and thermal stress. These conditions can alter the physical structure and chemical equilibrium of elastomeric stoppers. After reconstitution, these changes can enhance the migration rate of organic compounds from the stopper into the drug product. This makes lyophilized formulations more susceptible to leachable-related contamination compared to standard liquid systems.
A delay column is used to separate system-generated background signals from actual sample-derived analytes. It retains trace nitrosamines originating from solvents, tubing, or pump components, causing them to elute at different retention times. This separation prevents overlap between background contamination and target compounds. As a result, it significantly improves analytical accuracy and reliability in ultra-trace detection workflows.
The CPCA is used when direct toxicological data for a nitrosamine impurity are not available. It evaluates molecular structure using predictive parameters such as functional groups, electronic distribution, and steric effects. Based on these features, compounds are assigned to defined potency categories. Each category corresponds to a calculated acceptable intake limit that ensures patient safety in the absence of compound-specific data.
Reference:
- European Medicines Agency. (n.d.). Nitrosamine impurities. https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/referral-procedures-human-medicines/nitrosamine-impurities
- Wichitnithad, W., Nantaphol, S., Noppakhunsomboon, K., & Rojsitthisak, P. (2023). An update on the current status and prospects of nitrosation pathways and possible root causes of nitrosamine formation in various pharmaceuticals. Saudi Pharmaceutical Journal, 31(2), 295–311. https://doi.org/10.1016/j.jsps.2022.12.010
- U.S. Food and Drug Administration. (2024, September). Control of nitrosamine impurities in human drugs: Guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/control-nitrosamine-impurities-human-drugs
- U.S. Food and Drug Administration. (2024, September). Control of nitrosamine impurities in human drugs: Guidance for industry. https://www.fda.gov/media/170794/download
- U.S. Food and Drug Administration. (2023, August). Recommended acceptable intake limits for nitrosamine drug substance-related impurities (NDSRIs): Guidance for industry. https://www.fda.gov/media/170794/download
- United States Pharmacopeia. (2020, November). Highlights of General Chapter <1469> Nitrosamine impurities [Presentation]. https://www.usp.org/sites/default/files/usp/document/stakeholder-forum/pnp/highlights-of-1469-nitrosamine-impurities.pdf
- United States Pharmacopeia. (2022, April 4). Nitrosamines published limits – Reference. Nitrosamines Exchange. https://nitrosamines.usp.org/t/nitrosamines-published-limits-reference/2139

