
Introduction
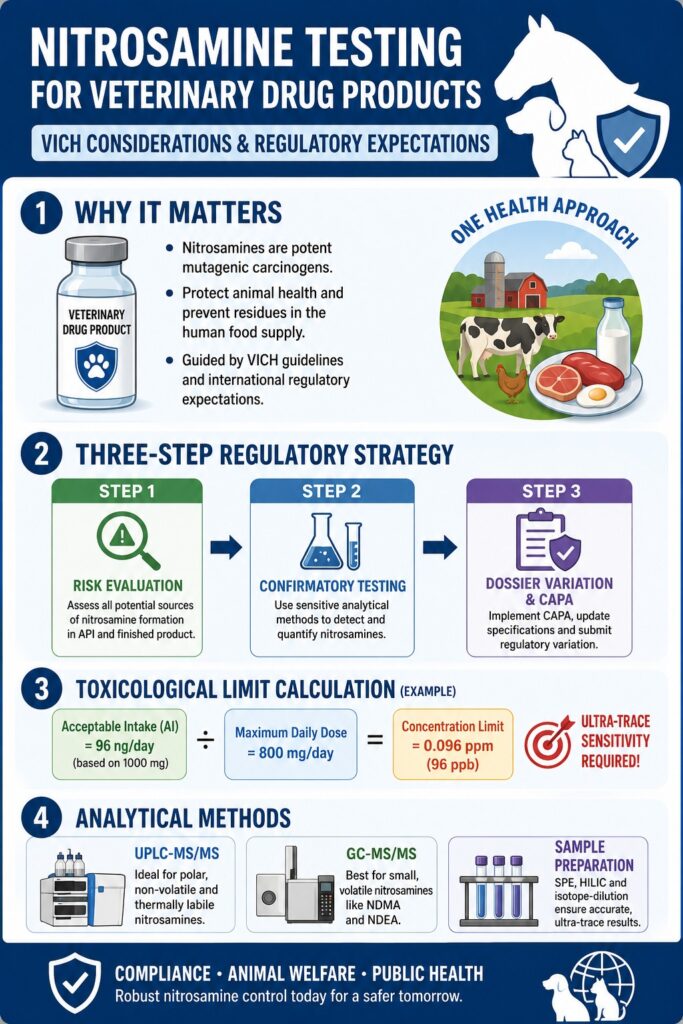
Nitrosamine Testing for Veterinary Drug Products is a critical analytical quality control process used to identify and quantify mutagenic N-nitroso impurities in veterinary pharmaceuticals. Its primary objective is to safeguard animal health while minimizing the risk of harmful residues entering the human food supply. This testing strategy is guided by internationally harmonized regulatory frameworks and regional requirements that recognize nitrosamines as highly potent carcinogenic compounds.
Access the foundational science: Download our complete technical brief defining the chemical composition of nitrosamines and their specific toxicological risks to veterinary health at Learn What Are Nitrosamines and Their Impact.
Global regulatory authorities first recognized the widespread risk of N-nitrosamine contamination in mid-2018 after detecting N-nitrosodimethylamine (NDMA) in chemically synthesized human antihypertensive medicines containing “sartan” active ingredients. Although the initial regulatory response focused on human medicinal products, attention has increasingly shifted toward veterinary pharmaceuticals. This evolution aligns with the modern One Health approach, which emphasizes the close interconnection between animal health, human health, and food production systems.
Veterinary medicinal products present distinct exposure routes, species-specific toxicokinetic characteristics, and the possibility of residue accumulation throughout the food chain. Addressing these challenges requires specialized expertise from experienced contract development and manufacturing organizations (CDMOs). ResolveMass Laboratories Inc. assists veterinary pharmaceutical manufacturers by delivering compliant ultra-trace nitrosamine testing solutions that are developed to meet stringent global regulatory expectations.
Partner with a specialized laboratory to ensure global regulatory alignment: Learn more about Outsourcing Nitrosamine Testing to an Expert CRO.
Article Summary:
- Nitrosamine testing is essential for veterinary medicines because these impurities are highly potent mutagenic carcinogens that can affect animal health and, in food-producing species, pose a potential risk to consumers through residue exposure.
- Standard VICH impurity thresholds are not sufficient for nitrosamines. Instead, manufacturers must follow dedicated mutagenic impurity guidance that requires ultra-low limits based on toxicological risk rather than conventional impurity reporting criteria.
- Acceptable nitrosamine limits are calculated using toxicological data and product exposure. Factors such as maximum daily dose, target animal species, treatment duration, and food safety considerations determine the allowable concentration for each veterinary product.
- Nitrosamines may form through multiple manufacturing-related pathways, including reactions between amines and nitrosating agents, contaminated raw materials, recovered solvents, water impurities, and unfavorable processing conditions such as acidic environments or elevated temperatures.
- Regulatory authorities expect a structured three-step risk management approach: perform a comprehensive risk assessment, conduct confirmatory testing with validated analytical methods, and implement corrective actions with regulatory dossier updates when necessary.
- Advanced analytical technologies are required to detect nitrosamines at trace levels. Techniques such as UPLC-MS/MS, GC-MS/MS, effective sample preparation, and isotope-dilution methods provide the sensitivity and accuracy needed for reliable regulatory compliance.
- Continuous lifecycle management is critical for maintaining compliance. Manufacturers must evaluate process changes, update Certificates of Suitability (CEP) and marketing authorizations when required, and integrate ongoing nitrosamine control strategies into routine quality systems to protect animal welfare and public health.
VICH Impurity Framework and Mutagenic Specifics
The VICH guideline framework establishes internationally harmonized requirements for reporting, identifying, and qualifying organic impurities in veterinary medicinal products. However, these conventional impurity thresholds are not applicable to highly mutagenic carcinogens such as nitrosamines. Instead, nitrosamines are regulated under dedicated non-clinical safety guidance that requires these impurities to be controlled at exceptionally low concentrations.
The International Cooperation on Harmonisation of Technical Requirements for Registration of Veterinary Medicinal Products (VICH) defines impurity control principles through two core guidelines:
- VICH GL10: Impurities in New Veterinary Drug Substances
- VICH GL11: Impurities in New Veterinary Medicinal Products
According to VICH GL10, organic impurities present in new veterinary drug substances are classified into categories such as starting materials, synthesis by-products, intermediates, degradation products, reagents, ligands, and catalysts. VICH GL11 further establishes reporting thresholds (typically 0.3%) and identification thresholds (typically 1.0%) for degradation products detected during stability studies of veterinary medicinal products.
Nevertheless, both VICH GL10 and VICH GL11 emphasize that analytical procedures should be specifically designed to detect impurities capable of producing significant toxicological effects even when present below conventional identification thresholds. Since N-nitrosamines are recognized as highly potent mutagenic impurities, they cannot be adequately assessed using standard impurity reporting limits.
To address this regulatory limitation, the European Medicines Agency (EMA) together with the Committee for Veterinary Medicinal Products (CVMP) introduced the Guideline on Assessment and Control of DNA Reactive (Mutagenic) Impurities in Veterinary Medicinal Products (EMA/CVMP/SWP/377245/2016). This guidance incorporates the Threshold of Toxicological Concern (TTC) concept into veterinary medicine, ensuring that nitrosamines and other DNA-reactive contaminants remain below concentrations associated with a meaningful carcinogenic risk.
Understand the core regulatory principles governing DNA-reactive contaminants by reading our guide on Genotoxic Impurity Testing and ICH M7 Compliance.
Toxicological Calculations and Species-Specific Limits
The maximum permissible concentration of a nitrosamine in a veterinary medicinal product is determined by dividing the toxicological Acceptable Intake (AI) by the Maximum Daily Dose (MDD) of the active pharmaceutical ingredient. These calculations differ considerably depending on target animal species, body weight, anticipated lifetime exposure, and food safety considerations related to human consumption.
For companion animals, including dogs and cats, toxicological assessments are based on a default target animal Threshold of Toxicological Concern (TTC) of 0.025 μg/kg body weight/day. The concentration limit (C) expressed in parts per million (ppm) is calculated using the following equation:
C = Acceptable Daily Intake (ng/day) ÷ Maximum Daily Dose (mg/day)
To illustrate the practical application of this calculation, consider a veterinary medicinal product with a Maximum Daily Dose (MDD) of 800 mg/day. If toxicological evaluation establishes a compound-specific Acceptable Intake (AI) of 96 ng/day based on a reference dose of 1000 mg, the adjusted AI (X) for an 800 mg/day dose is calculated as follows:
X = (96 ng/day × 800 mg/day) ÷ 1000 mg/day = 76.8 ng/day
Applying the concentration limit equation gives:
C = 76.8 ng/day ÷ 800 mg/day = 0.096 ppm
This calculated limit of 0.096 ppm (equivalent to 96 ppb) clearly demonstrates the extremely high analytical sensitivity required during raw material qualification and routine screening.
For veterinary medicinal products intended for short-term administration, a Less-Than-Lifetime (LTL) adjustment may be scientifically justified using the default target animal lifespan assumptions established by the CVMP. In contrast, medicinal products intended for food-producing animals require substantially more conservative safety limits. Because consumers receive no therapeutic benefit from veterinary drug residues present in meat, milk, eggs, or honey, mutagenic impurities in these products are controlled according to a consumer residue safety threshold of 0.0025 μg/kg body weight/day.
| Target Species / Category | Baseline Weight | Expected Treatment Lifespan Basis | Acceptable Daily Intake Basis | Regulatory Reference |
|---|---|---|---|---|
| Small Dogs (<10 kg) | <10 kg | 20 Years (LTL Baseline) | 0.025 μg/kg body weight/day | EMA/CVMP/SWP/32272/2022 |
| Medium Dogs (10–39 kg) | 10–39 kg | 15 Years (LTL Baseline) | 0.025 μg/kg body weight/day | EMA/CVMP/SWP/32272/2022 |
| Large Dogs (≥40 kg) | ≥40 kg | 10 Years (LTL Baseline) | 0.025 μg/kg body weight/day | EMA/CVMP/SWP/32272/2022 |
| Cats | Standardized | 18 Years (LTL Baseline) | 0.025 μg/kg body weight/day | EMA/CVMP/SWP/32272/2022 |
| Honeybees (Colony) | 15 kg (Colony Default) | Treatment Cycle Duration | Colony-wide 0.375 μg/colony/day | EMA/CVMP/SWP/32272/2022 |
| Food-Producing Species | Variable by Species | Slaughter Weight Target | Consumer Safety Limit: 0.0025 μg/kg body weight/day | EMA/CVMP/SWP/32272/2022 |
Risk Factors and Chemical Mechanisms of Nitrosamine Formation
The formation of nitrosamines in veterinary medicinal products is predominantly driven by chemical reactions between secondary, tertiary, or quaternary amines and nitrosating agents under favorable acidic or elevated-temperature conditions. Identifying these potential reaction pathways requires a thorough evaluation of the synthetic manufacturing process, recovered solvent systems, reagent quality, and possible contributions from packaging-related leachables.
Amine and Nitrosating Agent Interaction Chemistry
The principal pathway responsible for nitrosamine generation involves the reaction of susceptible amine-containing compounds with reactive nitrosating species. During active pharmaceutical ingredient (API) manufacturing, these reactive components may originate from multiple process-related sources, including:
- Vulnerable Amines: Secondary or tertiary amine functionalities present within the API structure itself, synthesis intermediates, process reagents, catalysts, or commonly used solvents such as dimethylamine and triethylamine.
- Nitrosating Agents: Sodium nitrite (NaNO₂), alkyl nitrites, nitrogen oxides (NOₓ), or nitric acid contaminated with nitrous acid or dinitrogen tetroxide.
- Acidic and Thermal Conditions: Acidic environments combined with elevated temperatures promote the conversion of nitrites into nitrous acid. Subsequent dehydration reactions generate highly reactive nitrosating intermediates, including the nitrosyl cation (NO⁺) and dinitrogen trioxide (N₂O₃), which readily react with susceptible amines to produce N-nitrosamines.
Amine (R₁R₂NH) + Nitrosating Agent (NO⁺) → N-Nitrosamine (R₁R₂N-N=O) + H⁺
(Reaction proceeds under acidic conditions and/or elevated temperature.)
One significant but frequently underestimated source of nitrosamine contamination during veterinary pharmaceutical manufacturing is the reuse of recovered solvents, catalysts, and processing reagents. When solvent recovery operations are performed in non-dedicated manufacturing facilities, trace quantities of residual amines originating from previous production campaigns may remain in the recovered materials. These residual amines can subsequently react with nitrite-containing contaminants within solvent streams, leading to the formation of persistent nitrosamines that may be transferred into later API manufacturing batches, creating an ongoing cross-contamination risk.
Review detailed chemical degradation pathways by visiting Nitrosamine Formation Pathways in API Synthesis.
Matrix-Specific Raw Material Concerns
Beyond synthesis-related chemistry, raw materials and environmental factors can also introduce nitrosating agents into veterinary drug products. Excipients commonly incorporated into solid oral dosage forms—including microcrystalline cellulose, lactose, and calcium hydrogen phosphate—may contain trace levels of nitrites generated during their manufacturing processes. Although these impurities are typically present in very small quantities, they can contribute to nitrosamine formation under favorable reaction conditions.
Water quality is another important consideration during API manufacturing. Analytical studies have shown that purified water generally contains less than 0.1 ppb of nitrite, whereas potable water may contain concentrations exceeding 3 ppb. Consequently, comprehensive monitoring and verification of water quality are essential components of process validation and overall nitrosamine risk management.
Differentiate between process-driven contamination and container closure risks: Read about the Difference Between Nitrosamine Impurities and Nitrosamine Leachables.
Regulatory Lifecycles and the Three-Step Strategy
Regulatory agencies expect manufacturers to manage nitrosamine risk through a structured three-step lifecycle approach consisting of risk evaluation, confirmatory testing, and marketing authorization variation. This systematic process ensures that manufacturers thoroughly assess potential sources of contamination, verify impurity levels using validated analytical methods, and update regulatory submissions whenever necessary.

Step 1: Quality Risk Evaluation
The first stage requires Marketing Authorization Holders (MAHs) to perform a comprehensive quality risk assessment covering both chemically synthesized active pharmaceutical ingredients and finished veterinary medicinal products. This evaluation should examine every aspect of the manufacturing process, including synthetic routes, raw material suppliers, excipient sources, recovered materials, manufacturing equipment, and primary packaging components. The completed assessment must be documented and submitted to the appropriate regulatory authorities as part of the manufacturer’s compliance obligations.
Find out if your specific portfolio requires mandatory documentation: Read Do All Drugs Need a Nitrosamine Risk Assessment?.
Step 2: Confirmatory Testing
When the initial risk assessment identifies a potential pathway for nitrosamine formation or contamination, confirmatory analytical testing becomes mandatory. This phase requires the finished veterinary medicinal product to be analyzed using validated, highly sensitive analytical methods capable of detecting nitrosamines at ultra-trace concentrations. The objective is to determine whether nitrosamines are present and accurately quantify their concentration.
If analytical testing confirms that nitrosamine concentrations exceed the applicable acceptable intake limits, or if a previously unidentified nitrosamine is detected, the Marketing Authorization Holder must promptly notify the relevant competent regulatory authorities in accordance with applicable reporting requirements.
Evaluate your data against established thresholds: Discover the nuances of Nitrosamine Alert Limits vs. Action Limits.
Step 3: Dossier Variation and CAPA Implementation
When confirmatory testing verifies the presence of nitrosamines, manufacturers are required to implement a comprehensive Corrective and Preventive Action (CAPA) program. Corrective actions may involve modifications to the manufacturing process, reformulation of the finished dosage form to eliminate conditions favorable for nitrosamine formation, improvements in raw material specifications, or enhanced supplier qualification programs to reduce contamination risks.
To maintain regulatory compliance, manufacturers must submit a formal variation to their marketing authorization, updating product specifications and incorporating routine nitrosamine control strategies into batch release testing. Regulatory guidance generally requires these CAPA activities to be completed within a maximum period of three years following establishment of the applicable acceptable intake limits.
Explore risk mitigation strategies if changes are required: Learn more about our Nitrosamine Reformulation Strategy Services.
Analytical Methodology for Nitrosamine Testing for Veterinary Drug Products
Highly sensitive chromatographic separation techniques coupled with tandem mass spectrometry represent the current analytical gold standard for detecting trace-level nitrosamines in complex veterinary medicinal products. These advanced analytical platforms provide the selectivity and sensitivity necessary to overcome significant matrix interference arising from complex veterinary formulations while generating scientifically robust data suitable for regulatory submissions.
Chromatography and Tandem Mass Spectrometry Platforms
Because regulatory acceptable intake limits for nitrosamines are typically established within the parts-per-billion (ppb) range, analytical testing requires highly selective and extremely sensitive instrumentation. The principal analytical platforms used for Nitrosamine Testing for Veterinary Drug Products include:
- Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS): This platform is particularly well suited for analyzing polar, non-volatile, and thermally unstable nitrosamines, as well as structurally complex Nitrosamine Drug Substance-Related Impurities (NDSRIs).
- Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS): This technique is ideally suited for small, volatile nitrosamine compounds such as N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) that can be efficiently vaporized without decomposition.
To promote analytical consistency and method reliability, the European Pharmacopoeia General Chapter 2.5.42 (N-Nitrosamines in Active Substances) provides standardized analytical approaches categorized as Method A (LC-MS/MS), Method B (GC-MS), and Method C (GC-MS/MS). These validated approaches serve as reliable starting points for laboratory method development and validation.
Discover the critical differences between complex and simple configurations: Check out NDSRIs vs. Simple Nitrosamines.
Overcoming Matrix Interference through Sample Preparation
Veterinary medicinal products often present significant analytical challenges because of their highly diverse formulation matrices, which may contain elevated concentrations of lipids, flavoring agents, preservatives, and polymeric stabilizers. Effective sample preparation is therefore essential to isolate trace nitrosamine analytes while minimizing matrix-related interference. Commonly applied sample preparation strategies include:
- Solid-Phase Extraction (SPE): Concentrates target nitrosamine analytes while removing the majority of excipient-derived matrix components, thereby reducing analytical background and significantly improving detection sensitivity.
- Hydrophilic Interaction Chromatography (HILIC): Utilized as either a sample clean-up technique or an analytical separation strategy to improve retention characteristics and chromatographic peak shape for highly polar compounds present in complex liquid and semi-solid formulations.
- Isotope-Dilution Quantification: Employs carbon-13 (¹³C) or deuterium-labeled (d) internal standards to compensate for analyte recovery losses, ion suppression, and matrix-induced signal enhancement, thereby improving quantitative accuracy.
These analytical methodologies must be validated in accordance with VICH GL1 and VICH GL2 requirements. Critical validation parameters include specificity, linearity, accuracy, precision, limit of detection (LOD), and limit of quantification (LOQ), ensuring that the resulting analytical data are scientifically robust and fully acceptable for regulatory review.
Optimize your validation workflow with compliant protocols: Explore our Nitrosamine Method Development and Validation Services.
Dossier Integration and CEP Variation Management
Certificates of Suitability (CEPs) that include established nitrosamine limits must be carefully evaluated by veterinary Marketing Authorization Holders to confirm that the specified limits remain appropriate for the intended target species, dosage regimen, and duration of treatment. Any modification to the active pharmaceutical ingredient manufacturing process or updates to an existing CEP may require the prompt submission of regulatory variations in accordance with the applicable classification procedures.
The Role of CEP 2.0 in Veterinary Dossiers
The Certificate of Suitability (CEP) procedure administered by the European Directorate for the Quality of Medicines & HealthCare (EDQM) is widely recognized as a mechanism for demonstrating that active pharmaceutical ingredients comply with the relevant European Pharmacopoeia monographs. Under the CEP 2.0 framework, when a nitrosamine specification is included within the CEP, that limit is regarded as acceptable for general pharmacopoeial compliance.
Nevertheless, the veterinary Marketing Authorization Holder (MAH) retains ultimate responsibility for confirming that the CEP-defined nitrosamine limit remains appropriate for the finished veterinary medicinal product. This assessment requires consideration of the target animal species, maximum daily dose, and anticipated duration of exposure. If the nitrosamine limit specified within the CEP exceeds the calculated safety threshold for the intended veterinary application, the MAH must establish more stringent product-specific release specifications within the regulatory dossier.
Post-Approval Variation Strategies and Nitrosamine Testing for Veterinary Drug Products
Whenever an active pharmaceutical ingredient manufacturer modifies the synthesis process or introduces manufacturing changes that alter the impurity profile covered by the CEP, the Marketing Authorization Holder must update the product’s regulatory dossier accordingly. These updates are generally classified under the following European regulatory variation categories:
- Type IA Administrative Updates: Applicable when administrative information, such as the manufacturer’s name or physical address, is revised without affecting manufacturing processes or approved production locations.
- Type IB or Type II Variation Pathways: Required when a revised or newly issued CEP introduces or modifies an existing nitrosamine specification. Such variations should be submitted promptly under Classification Scope Q.III.1, ensuring that all quality- and safety-related changes are formally incorporated into the marketing authorization.
To maintain full regulatory compliance, the Qualified Person (QP) declaration contained within Module 1 (or VNeeS Part 1) should identify every active manufacturing location, intermediate synthesis site, and physical processing facility—including micronization and lyophilization sites—to ensure complete transparency and oversight throughout the pharmaceutical supply chain.
Ensure compliance before final market distribution: Review the Nitrosamine Batch Release Testing Requirements.
Conclusion
Implementing a comprehensive strategy for Nitrosamine Testing for Veterinary Drug Products plays a fundamental role in achieving global regulatory compliance while safeguarding both animal welfare and public health. By following a structured three-step lifecycle management approach, veterinary pharmaceutical manufacturers can systematically identify potential sources of nitrosamine formation, perform highly sensitive confirmatory testing, and maintain scientifically robust regulatory submissions throughout the product lifecycle.
As worldwide regulatory expectations continue to evolve under the One Health framework, manufacturers must rely on advanced trace-level chromatographic and mass spectrometric technologies capable of meeting increasingly stringent compliance requirements. Collaborating with an experienced analytical laboratory enables comprehensive characterization of complex veterinary formulations while supporting the protection of target animal species and the safety of the human food supply.
The mass spectrometry scientists at ResolveMass Laboratories Inc. possess the advanced analytical instrumentation, regulatory knowledge, and method validation expertise required to support compliance with these continually evolving global expectations. Veterinary pharmaceutical developers and manufacturers can simplify their nitrosamine risk assessment strategies and strengthen regulatory readiness by initiating an analytical consultation through our Contact Us page.
Frequently Asked Questions
For companion animals such as dogs and cats, the default Threshold of Toxicological Concern (TTC) for mutagenic impurities is 0.025 μg/kg body weight per day. This value serves as the toxicological reference point when establishing acceptable exposure levels during veterinary risk assessments. It helps ensure that long-term exposure to trace mutagenic contaminants remains within a level considered to present a minimal carcinogenic risk.
Veterinary medicines intended for food-producing animals are assessed using a highly conservative consumer safety approach. The acceptable exposure limit is based on a Threshold of Toxicological Concern (TTC) of 0.0025 μg/kg body weight per day, which is designed to protect people consuming meat, milk, eggs, honey, or other animal-derived products. This approach minimizes the possibility of mutagenic residues entering the human food chain.
The CVMP provides default lifespan assumptions that support Less-Than-Lifetime (LTL) exposure calculations for companion animals. These include 20 years for small dogs, 15 years for medium-sized dogs, 10 years for large dogs, and 18 years for cats. These standardized values are used to estimate cumulative exposure during shorter treatment periods and to establish scientifically justified acceptable intake limits.
In toxicological evaluations, a honeybee colony is considered a single biological unit or “superorganism” rather than assessing individual bees separately. A default colony weight of 15 kg is used together with the target animal TTC of 0.025 μg/kg body weight per day to calculate the overall acceptable exposure limit. This approach results in a colony-wide acceptable intake of 0.375 μg per colony per day, ensuring protection of the entire colony.
The two principal analytical techniques used for veterinary nitrosamine analysis are Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) and Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS). These advanced platforms provide the high sensitivity and selectivity required to detect nitrosamines at parts-per-billion concentrations. They are particularly effective for analyzing complex veterinary formulations containing challenging excipient matrices.
Both excipients and manufacturing water can contribute to nitrosamine formation if they contain trace amounts of nitrites. Certain pharmaceutical excipients may introduce residual nitrites from their production processes, while potable water generally contains higher nitrite concentrations than purified water. Evaluating these materials helps manufacturers identify potential sources of contamination and implement appropriate controls during formulation and manufacturing.
Although a Certificate of Suitability (CEP) may include an approved nitrosamine specification, the veterinary Marketing Authorization Holder remains responsible for determining whether that limit is appropriate for the finished medicinal product. This assessment considers factors such as the maximum daily dose, treatment duration, and target animal species. If the CEP limit does not adequately protect the intended veterinary population, more stringent product-specific release limits should be established within the regulatory dossier.
When confirmatory testing identifies nitrosamine levels that exceed established acceptable limits, manufacturers are expected to implement a comprehensive Corrective and Preventive Action (CAPA) program. This may involve manufacturing process modifications, raw material improvements, or formulation changes to reduce nitrosamine formation. Regulatory guidance generally expects these corrective measures to be fully implemented within three years after publication of the applicable acceptable intake limits.
When a revised Certificate of Suitability introduces or updates a nitrosamine specification, the associated marketing authorization must also be updated to reflect the change. Depending on the nature of the revision, manufacturers are typically required to submit a Type IB or Type II variation under the applicable Q.III.1 classification. Prompt submission ensures that product quality specifications remain aligned with current regulatory and safety requirements.
Reference:
- European Medicines Agency. (2020). Guideline on assessment and control of DNA reactive (mutagenic) impurities in veterinary medicinal products (EMA/CVMP/SWP/377245/2016). https://www.ema.europa.eu/en/assessment-control-dna-reactive-mutagenic-impurities-veterinary-medicinal-products-scientific-guideline
- European Medicines Agency. (n.d.). Nitrosamine impurities in specific medicines. https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/pharmacovigilance-post-authorisation/referral-procedures-human-medicines/nitrosamine-impurities/nitrosamine-impurities-specific-medicines
- U.S. Food and Drug Administration. (2007). Guidance for Industry #92: Impurities in new veterinary drug substances (Revision) (VICH GL10(R)). https://www.fda.gov/media/70365/download
- Health Canada. (2022, April 4). Guidance on nitrosamine impurities in medications: Evaluating and managing the risks of N-nitrosamine impurities in human pharmaceutical, biological and radiopharmaceutical products. Government of Canada. https://www.canada.ca/content/dam/hc-sc/documents/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities/medications-guidance/guidance-nitrosamine%20impurities-medications.pdf
- European Medicines Agency. (2020, June 23). Lessons learnt from presence of N-nitrosamine impurities in sartan medicines (EMA/526934/2019). https://www.ema.europa.eu/en/documents/report/lessons-learnt-presence-n-nitrosamine-impurities-sartan-medicines_en.pdf
- European Medicines Agency. (n.d.). Nitrosamine impurities: Guidance for marketing authorisation holders. European Medicines Agency. https://www.ema.europa.eu/en/human-regulatory-overview/post-authorisation/pharmacovigilance-post-authorisation/referral-procedures-human-medicines/nitrosamine-impurities/nitrosamine-impurities-guidance-marketing-authorisation-holders
- European Medicines Agency. (n.d.). Quality guidelines for non-immunological veterinary medicinal products. European Medicines Agency. https://www.ema.europa.eu/en/veterinary-regulatory-overview/research-development-veterinary-medicines/scientific-guidelines-veterinary-medicines/quality-guidelines/quality-guidelines-non-immunologicals

