
Introduction: Advanced Advancements in Peptide Impurity Identification and Characterization Services
Peptide Impurity Identification and Characterization Services are specialized analytical solutions developed to isolate, identify, and structurally confirm trace-level organic impurities and degradation products present in peptide drug substances and finished pharmaceutical products. These services generate comprehensive, high-resolution analytical data that are essential for demonstrating active pharmaceutical ingredient (API) sameness, elucidating forced degradation pathways, and satisfying the rigorous compliance requirements established by global regulatory authorities. In modern pharmaceutical development, these services play a pivotal role in supporting the transition from recombinant originator products to generic synthetic peptide alternatives, a process governed by stringent international quality standards.
Within today’s biopharmaceutical industry, implementing robust Peptide Impurity Identification and Characterization Services is fundamental to ensuring both patient safety and therapeutic equivalence. The global peptide therapeutics market continues to experience significant growth, largely driven by the clinical success of complex peptide molecules such as glucagon-like peptide-1 (GLP-1) receptor agonists, including semaglutide, liraglutide, and tirzepatide. Unlike conventional small-molecule drugs, peptides are structurally complex polymers whose biological performance and safety characteristics are closely associated with their higher-order conformations and impurity profiles. The United States Food and Drug Administration (FDA), along with other international regulatory agencies, imposes highly stringent requirements for controlling peptide-related impurities. According to the FDA’s final guidance issued in May 2021, any peptide-related impurity present in a proposed generic synthetic peptide at a concentration of ≥ 0.10% must be structurally identified and appropriately qualified. As a result, partnering with an experienced analytical organization equipped with advanced scientific expertise and state-of-the-art instrumentation, such as ResolveMass Laboratories Inc., has become a mission-critical decision for biotechnology and pharmaceutical companies worldwide.
Get Expert Support: Partner with an experienced team for your drug development needs. Explore our comprehensive GLP-1 Peptide Impurity Characterization Services to ensure global regulatory compliance.
Share via:
Article Summary:
- Peptide impurity identification and characterization is essential for ensuring the safety, quality, and regulatory compliance of peptide-based pharmaceuticals by detecting and confirming trace-level process- and degradation-related impurities.
- Regulatory agencies, including the FDA, require rigorous impurity profiling, with peptide-related impurities at or above 0.10% in generic synthetic peptides requiring structural identification and scientific justification to demonstrate therapeutic equivalence.
- Process-related impurities mainly originate during peptide synthesis and purification, including amino acid deletions, insertions, truncated sequences, protecting group adducts, and racemization, all of which can affect product purity and manufacturing consistency.
- Degradation-related impurities develop during storage through chemical reactions such as oxidation, deamidation, aspartimide formation, hydrolysis, and disulfide scrambling, making comprehensive stability studies critical for product development.
- Advanced analytical technologies such as high-resolution mass spectrometry (HRMS), two-dimensional liquid chromatography (2D-LC), and complementary spectroscopic techniques enable accurate identification of structurally similar, co-eluting, and isomeric peptide impurities.
- Reliable impurity quantification relies on Relative Response Factor (RRF) calculations, while integrated approaches such as qNMR-LC improve measurement accuracy when impurity reference standards are unavailable.
- Comprehensive impurity characterization supports successful drug development by helping pharmaceutical companies meet global regulatory expectations, reduce development risks, accelerate approvals, and ensure the production of safe, high-quality peptide therapeutics.

Regulatory Thresholds and the Critical Role of Peptide Impurity Identification and Characterization Services
Regulatory requirements specify that peptide-related impurities in generic synthetic drug products exceeding a concentration threshold of ≥ 0.10% must undergo comprehensive structural identification and characterization to demonstrate therapeutic equivalence with the reference listed drug. Proper identification and qualification of impurities are essential for minimizing the potential risks of increased immunogenicity and toxicity during clinical use. These regulatory expectations are particularly stringent for synthetic peptide generics intended to demonstrate equivalence to reference listed drugs (RLDs) produced through recombinant deoxyribonucleic acid (rDNA) technology.
To support Abbreviated New Drug Application (ANDA) submissions, the FDA has established detailed comparative evaluation requirements for five highly purified synthetic peptide drug products: glucagon, liraglutide, nesiritide, teriparatide, and teduglutide. The assessment of active pharmaceutical ingredient sameness encompasses multiple critical quality attributes, including the primary amino acid sequence, physicochemical characteristics, secondary and tertiary structural conformations, oligomerization and aggregation profiles, biological activity, and, most importantly, comparative impurity profiling.
The differences in regulatory thresholds across major regulatory frameworks are summarized below.
| Regulatory Parameter / Framework | European Pharmacopoeia (Ph. Eur.) Monograph 2034 | FDA Generic Synthetic Peptide Guidance (May 2021) |
|---|---|---|
| Identification Threshold | ≥ 0.5% (Impurities below this level are reported as unknown.) | ≥ 0.10% (All impurities at or above this limit must be identified and characterized.) |
| Qualification Threshold | ≥ 1.0% (Identified impurities up to this limit are accepted without additional qualification.) | ≤ 0.5% for any new specified peptide-related impurity |
| New Specified Impurities | Generally accepted within compendial monograph limits | Unacceptable if > 0.5%; if between 0.10% and 0.5%, a formal risk assessment is required. |
| Unspecified Impurity Limit | ≤ 0.5% [cite: 10] | Strictly less than 0.10% [cite: 12] |
Furthermore, the Threshold of Toxicological Concern (TTC) of 1.5 μg/day established under the International Council for Harmonisation (ICH) M7 guideline provides the basis for evaluating the safety of previously uncharacterized impurities. For therapeutic peptides administered at relatively low daily doses, this threshold offers considerable regulatory flexibility. For instance, teriparatide has a maximum daily dose of 20 μg/day. At this dosage, a related impurity limit of 1.0% corresponds to an individual patient exposure of only 0.2 μg/day. This exposure level remains substantially below the TTC value of 1.5 μg/day, illustrating how scientific evidence and toxicological justification can be effectively used to support regulatory submissions. Nevertheless, when a new specified peptide-related impurity is present exclusively in the generic synthetic product and not in the reference listed drug, with a concentration ranging between 0.10% and 0.5%, the applicant is required to perform a comprehensive immunogenicity risk assessment. This assessment typically includes in silico T-cell epitope mapping alongside cell-based in vitro studies to evaluate the potential for both innate and adaptive immune responses.
Ensure Full Compliance: Streamline your standard approvals by reading our detailed breakdown of the Regulatory Requirements for GLP-1 Peptide Characterization.
Molecular Taxonomy and Synthesis Mechanisms of Process-Related Impurities
Process-related peptide impurities arise directly during chemical synthesis, peptide cleavage, and purification processes. These impurities commonly include amino acid deletions, insertions, truncated peptide chains, and permanent protecting group adducts. Effective control of these impurities requires the establishment of stringent specifications for protected Fmoc-amino acid starting materials, together with careful optimization of peptide coupling conditions. Since these contaminants are carried into the crude peptide product, they frequently exhibit chromatographic behavior that is highly similar to that of the desired target peptide, making their separation particularly challenging.
Impact of Starting Material Specifications on Process Impurity Minimization
The impurity profile of protected Fmoc-amino acid starting materials has a direct influence on the crude purity of the synthesized peptide because even trace-level contaminants may trigger irreversible chain termination events or double amino acid insertions during synthesis. Consequently, establishing rigorous quality specifications for residual solvents, free amino acids, and dipeptide impurities is essential for minimizing downstream purification difficulties and ensuring consistent manufacturing performance.
To achieve highly reproducible peptide synthesis and maintain clean manufacturing profiles, protected Fmoc-amino acid starting materials should comply with stringent quality specifications.
| Starting Material Impurity Type | Synthesis Source & Mechanism | Target Specification Limit | Downstream Impact on Final Peptide |
|---|---|---|---|
| Residual Free Amino Acids | Incomplete protection during Fmoc-amino acid synthesis or premature Fmoc cleavage during storage. | ≤ 0.2% | Unprotected amino acids promote multiple insertions or undergo autocatalytic cleavage. |
| Dipeptide Contaminants (Fmoc-Xaa-Xaa-OH) | Formation occurs when the Fmoc introduction reagent reacts with previously generated Fmoc-amino acid. | ≤ 0.1% | Couples into the growing peptide chain, producing double amino acid insertion impurities. |
| β-Alanyl Species | Generated through ring-opening followed by molecular rearrangement of the Fmoc-OSu introduction reagent. | ≤ 0.1% | Results in direct incorporation of a β-alanine residue or replacement of the intended amino acid. |
| Acetate / Acetic Acid | Residual solvents, including ethyl acetate, remaining from crystallization and manufacturing operations. | ≤ 0.02% | Functions as a reactive chain-terminating capping agent, resulting in truncated peptide sequences. |
| Ethyl Acetate | Residual solvent originating from manufacturing and processing operations. | ≤ 0.5% | Gradually hydrolyzes during extended storage, generating reactive acetic acid. |
| Enantiomeric Impurity | Caused by incomplete enantiomeric separation or partial racemization during starting material synthesis. | ≥ 99.8% Enantiomeric Purity | Directly contributes to the formation of diastereomeric peptide impurities. |
| Overall HPLC Purity | Represents the cumulative presence of synthetic by-products. | ≥ 99.0% | Reduces crude peptide purity, thereby increasing the complexity of downstream purification processes. |
Deletion, Insertion, and Truncation Sequences
Deletion sequences (des-Xaa) and insertion sequences (endo-Xaa) are structural analogs that differ from the intended peptide by a mass shift corresponding to the molecular weight of the affected amino acid residue minus one water molecule (MW − 18.02 Da). Deletion impurities are generated when N-terminal Fmoc deprotection remains incomplete or when the subsequent amino acid coupling reaction fails to achieve complete conversion. As a result, unreacted amino groups persist into the following synthesis cycle, where they participate in subsequent coupling reactions, ultimately producing peptide sequences lacking a single amino acid residue.
Insertion impurities, in contrast, generally originate through three principal mechanisms:
- Starting Material Dipeptides: Direct incorporation of Fmoc-Xaa-Xaa-OH contaminants that are present within the protected starting materials.
- Premature Fmoc Loss: Premature removal of the backbone Fmoc protecting group during the coupling stage permits an additional coupling event involving the same activated amino acid. This unintended deprotection may be initiated by basic amine functionalities present on the peptide-resin, such as the ε-amine group of lysine or the secondary amine of proline, or by dimethylamine impurities present in the dimethylformamide (DMF) solvent.
- Incomplete Resin Washing: Insufficient washing of the peptide-resin following a coupling reaction allows excess activated amino acids to remain associated with the resin. During subsequent deprotection and coupling cycles, these residual activated species can react with the growing peptide chain, generating amino acid insertion impurities.
Truncated peptide sequences represent capped, incomplete peptide chains that result from irreversible chain-termination reactions during synthesis. Among the various contributing factors, the presence of trace quantities of acetic acid in starting materials is especially detrimental. Owing to its relatively low molecular weight, an acetic acid contamination level of only 0.1% by weight in a starting material corresponds to approximately 1 mol% contamination. Under conventional peptide synthesis conditions employing a five-fold excess of coupling reagents, even this trace amount of acetic acid can terminate as many as 5% of actively growing peptide chains during a single coupling cycle. In peptide sequences containing multiple sterically hindered amino acid residues, these cumulative chain-termination events may ultimately reduce the overall synthesis yield by as much as 15%.
Optimize Your Workflow: Review your structural criteria against industry baselines. Download our Peptide Characterization CRO Deliverables Checklist to manage process-related impurities effectively.
Protecting Group Adducts and Racemization Pathways
Incomplete removal of side-chain protecting groups, including tBu, Trt, and Pbf, at the end of solid-phase peptide synthesis results in the formation of covalently modified peptide-protecting group adducts. These adducts exhibit characteristic mass shifts, such as +56 Da for a residual tert-butyl group, and are particularly challenging to separate during analytical characterization. In addition to complicating purification, these modified species may induce functional cellular responses in biological systems, potentially producing false-positive outcomes during biological assays.
Enantiomeric racemization represents another significant process-related impurity pathway and primarily occurs during amino acid activation and fragment condensation steps throughout peptide synthesis. During activation of a protected amino acid or peptide fragment, the proton attached to the α-carbon becomes substantially more acidic. Under base-catalyzed conditions, such as deprotection using secondary amines like piperidine, the activated carboxyl group undergoes intramolecular cyclization, producing a five-membered oxazol-5(4H)-one intermediate. This intermediate readily loses the α-proton to generate a planar, resonance-stabilized enolate species, resulting in stereochemical inversion at the chiral center and subsequent incorporation of a D-amino acid instead of the native L-enantiomer. To minimize racemization, peptide synthesis protocols commonly employ coupling additives such as Oxyma Pure (ethyl 2-cyano-2-(hydroxyimino)acetate) or HOBt while maintaining reaction temperatures within the range of 0 °C to 50 °C.
Chemical Kinetics and Pathways of Degradation-Induced Impurities
Degradation-induced peptide impurities gradually develop throughout the product’s shelf life as a consequence of chemical reactions including aspartimide cyclization, deamidation, oxidation, and covalent polymerization. Forced degradation studies intentionally expose peptide molecules to harsh environmental conditions in order to identify these degradation pathways and support the development of stability-indicating analytical methods.
Aspartimide Cyclization and Reopening Kinetics
Aspartimide formation is among the most significant and challenging degradation mechanisms affecting peptides containing aspartate (Asp) or asparagine (Asn) residues. The reaction begins with deprotonation of the backbone amide nitrogen belonging to the adjacent C-terminal (n+1) amino acid residue. This nitrogen subsequently functions as an intramolecular nucleophile and attacks the neighboring β-carboxyl carbonyl group of the Asp residue, or the β-carboxamide group in the case of Asn. The resulting intramolecular cyclization forms a five-membered succinimide (aspartimide) intermediate while simultaneously releasing either one molecule of water (−18.02 Da) or one molecule of ammonia (−17.03 Da).
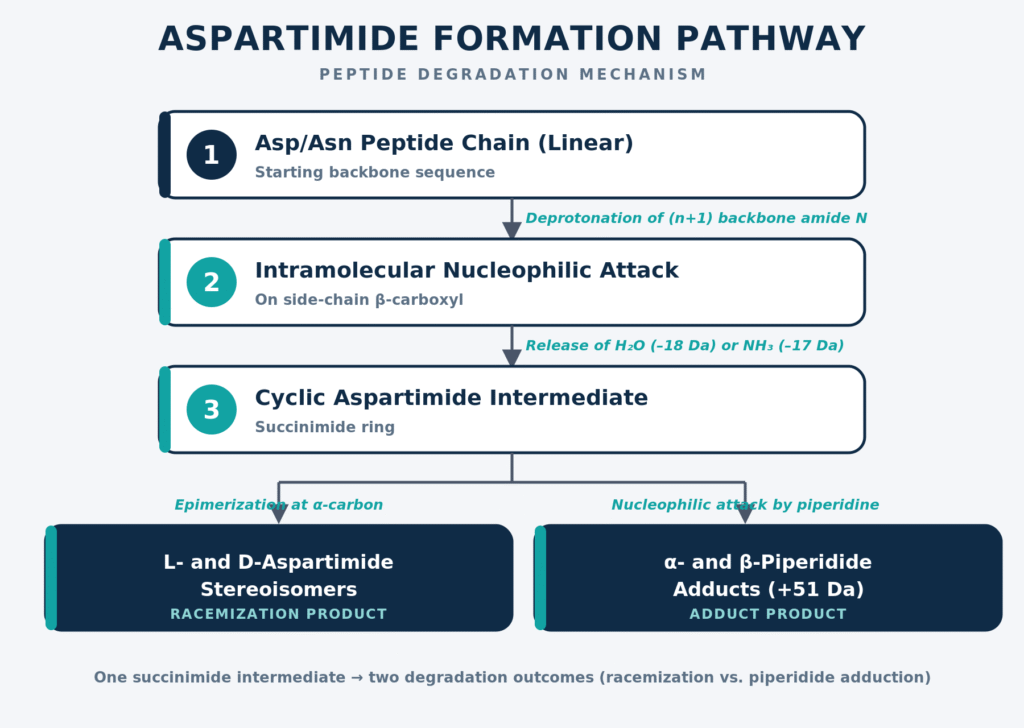
│ ▼ [Hydrolytic Ring-Opening by Water] Mixture of α-Asp and β-Asp (isoAsp) Peptides (approximately 1:3 ratio)
The cyclic aspartimide intermediate is highly prone to both racemization and nucleophilic ring-opening reactions. During hydrolysis, water may attack either carbonyl carbon of the succinimide ring, producing a mixture of the native linear α-aspartyl peptide and the isomerized β-aspartyl (isoaspartyl) peptide. The resulting products are typically generated in an approximate 1:3 ratio, favoring the thermodynamically more stable isoAsp form. Within Fmoc-based peptide synthesis, the secondary amine piperidine, used for Fmoc deprotection, may also attack the aspartimide ring to generate α- and β-piperidide adducts, producing a characteristic mass increase of +51 Da. Aspartimide formation proceeds particularly rapidly in Asp-Gly, Asp-Ala, and Asp-Ser sequence motifs because the adjacent (n+1) amino acid residue presents minimal steric hindrance. Common mitigation strategies include employing sterically bulky side-chain protecting groups such as CSY, MNI, or hydrazides, as well as backbone protection approaches using Dmb, Hmb, or GABA-Hmb to effectively shield the backbone amide nitrogen.
Deamidation and Hydrolysis Profiles
Deamidation is a major chemical degradation pathway that converts neutral asparagine (Asn) and glutamine (Gln) residues into the corresponding acidic amino acids, aspartate (Asp) and glutamate (Glu). This transformation introduces an additional negative charge while producing a characteristic mass increase of +0.98 Da. The reaction mechanism and resulting degradation products vary significantly depending on the surrounding pH conditions.
Acidic pH (pH < 3): Under strongly acidic conditions, deamidation proceeds through direct acid-catalyzed hydrolysis of the side-chain carboxamide group. This pathway bypasses formation of the cyclic imide intermediate and converts Asn directly into the native α-aspartyl peptide.
Neutral to Alkaline pH (pH > 5): At neutral and alkaline pH values, deamidation proceeds through an intramolecular cyclization mechanism. Deprotonation of the peptide backbone amide nitrogen promotes nucleophilic attack on the Asn side-chain carbonyl group, generating a cyclic glutarimide or succinimide intermediate. Subsequent hydrolytic opening of this cyclic intermediate produces a mixture of both α-aspartyl and β-aspartyl (isoaspartyl) peptide species.
Peptides are likewise susceptible to peptide bond hydrolysis, resulting in molecular fragmentation. Exposure to elevated temperatures or highly acidic or basic environments can promote cleavage of the peptide backbone, particularly at aspartyl peptide bonds. This degradation pathway generates lower-molecular-weight truncated peptide fragments that are readily identified through mass differences relative to the intact parent peptide.
Oxidation, Disulfide Scrambling, and β-Elimination
Oxidative degradation primarily affects sulfur-containing amino acids such as Methionine (Met) and Cysteine (Cys), as well as aromatic amino acid residues including Tryptophan (Trp), Tyrosine (Tyr), and Histidine (His). Oxidation of Methionine by reactive oxygen species results initially in the formation of methionine sulfoxide (+16 Da) and, under more aggressive oxidative conditions, methionine sulfone (+32 Da). These oxidative modifications alter the local physicochemical properties of the peptide, disrupting secondary structural organization and receptor-binding interactions.
For peptides containing multiple disulfide bonds, disulfide scrambling represents an important physical and chemical degradation mechanism. Under neutral or basic conditions, thiol-disulfide exchange reactions mediated by trace free thiols can rearrange native disulfide linkages, producing incorrectly paired conformers as well as covalent homodimers and heterodimers. These altered molecular species may trigger undesirable immune responses.
Additionally, under strongly basic conditions, Cysteine residues may undergo β-elimination of the protected sulfhydryl group, generating a highly reactive dehydroalanine (Dha) intermediate. During Fmoc-based peptide synthesis, this Dha intermediate readily reacts with piperidine to produce a 3-(1-piperidinyl)alanine impurity that exhibits a characteristic mass increase of +51 Da.
Ensure API Stability: Validate your shelf-life projections using state-of-the-art analytical configurations. Read more about GLP-1 Peptide Stability Analytical Methods to guard against long-term degradants.
Operational Execution of Peptide Impurity Identification and Characterization Services
Modern Peptide Impurity Identification and Characterization Services integrate high-resolution mass spectrometry (HRMS), multidimensional chromatography (2D-LC), and complementary spectroscopic techniques to achieve definitive structural characterization of co-eluting and isobaric peptide species. These orthogonal analytical platforms collectively generate comprehensive datasets capable of meeting the stringent chemical and physical characterization requirements established by regulatory agencies.
Resolution of Isobaric and Isomeric Peptide Species via Advanced Mass Spectrometry
Conventional bottom-up mass spectrometry workflows are unable to distinguish structural isomers such as Leucine and Isoleucine or modification isomers including isoglutamic acid because these species possess identical molecular masses. Accurate differentiation of these stereoisomeric substitutions requires advanced analytical strategies involving energy-resolved MS³ fragmentation of immonium ions together with comparative intensity profiling of product b and y ions.
Although high-resolution tandem mass spectrometry (HR-MS/MS) successfully confirms the amino acid composition of a peptide during primary sequence analysis, it cannot discriminate between Leucine (Leu) and Isoleucine (Ile) because both amino acids share the same monoisotopic mass and produce indistinguishable MS/MS backbone fragmentation patterns. To address this limitation, specialized Peptide Impurity Identification and Characterization Services utilize low-energy Electrospray Ionization (ESI) Trap MSⁿ instruments to isolate the m/z 86 immonium ion generated through source-induced collision dissociation. Subsequent MS³ fragmentation of this m/z 86 precursor produces a unique diagnostic pattern consisting of product ions at m/z 30, 44, and 69. The relative abundance of these ions enables definitive identification of Leu, Ile, or mixtures of both amino acids within digested peptide samples.
Similarly, distinguishing canonical aspartic acid (Asp) from isoaspartic acid (isoAsp), as well as glutamic acid (Glu) from isoglutamic acid (isoGlu), is of considerable analytical importance because these modifications alter the primary peptide backbone through insertion of an additional methylene group into the main chain. Although these isomeric species possess identical monoisotopic masses, backbone fragmentation efficiency changes significantly around the site of isomerization. By carefully evaluating the relative abundance of adjacent b and y fragment ions, analytical chemists can accurately identify and localize these structural modifications. Additional confirmation may be obtained by preparing reference standards using oxygen-18 (O¹⁸)-labeled water, which introduces an isomer-specific mass label that facilitates differentiation of the resulting carboxylic acid species.
Deploy Orthogonal Workflows: Advance beyond standard tandem MS limits. Learn how Multi-Attribute Monitoring (MAM) for Peptide Characterization tracks trace-level modifications simultaneously.
Chromatographic Orthogonality and Two-Dimensional Liquid Chromatography (2D-LC) Workflows
Two-dimensional liquid chromatography (2D-LC) significantly enhances chromatographic separation by integrating two independent chromatographic mechanisms in an online workflow, enabling efficient resolution of complex peptide mixtures and trace-level impurities. This analytical platform is particularly advantageous for online desalting applications, allowing heart-cut fractions collected from non-volatile pharmacopoeial mobile phases to be seamlessly transferred into volatile, mass spectrometry-compatible mobile phases for downstream analysis.

Achieving effective separation of co-eluting analytes requires the two chromatographic dimensions to exhibit a high degree of orthogonality, meaning that each dimension separates compounds according to different physicochemical properties. Although combining dissimilar stationary phases, such as C18 and Phenyl columns, provides only moderate orthogonality, substantially greater separation efficiency can be achieved by exploiting the ionizable properties of peptide molecules. Operating the first chromatographic dimension under basic conditions (for example, pH 10) and the second dimension under acidic conditions (for example, pH 2.6) creates a highly orthogonal separation environment with fractional coverage (f₍coverage₎) reaching approximately 68%. This approach provides superior chromatographic performance compared with alternative multidimensional configurations, including Strong Cation Exchange coupled with Reversed-Phase chromatography (SCX-RP) and Hydrophilic Interaction Liquid Chromatography coupled with Reversed-Phase chromatography (HILIC-RP).
When analyzing complex mixtures containing oxidation or deamidation isomers, Size Exclusion Chromatography (SEC) may also be integrated with Electron Transfer Dissociation (ETD) mass spectrometry. In this configuration, SEC causes modification isomers originating from the same peptide sequence to co-elute, enabling accurate relative quantification of all isomeric variants from a single ETD MS/MS spectrum.
Resolve Co-eluting Isomers: Combine high-resolution structural methods for ultimate certainty. Find out how we deploy 2D NMR for Peptide Characterization alongside multi-dimensional LC arrays.
Molecular Mapping of Specific Therapeutic Peptides and Their Impurities
The unique amino acid sequences of therapeutic peptides make each molecule susceptible to distinct process-related and degradation-related modifications. Therefore, comprehensive characterization studies must identify and map these sequence-specific alterations, including modifications such as oxidation at methionine-15 in teriparatide or deletion of serine-8 in liraglutide.
The sequence-specific deletions, insertions, oxidation sites, and additional functional group modifications observed in several important therapeutic peptides are summarized below.
| Peptide Common Name | Complete Amino Acid Sequence & Key Modifications | Characteristic Deletion / Insertion Sites | Confirmed Oxidation & Hydrolysis Sites | Other Functional Group Modifications / Isomerizations |
|---|---|---|---|---|
| Carbetocin | XYIQNCPLG (Modifications: X = Butanoic acid, Tyr-2 = Me-Tyr, Gly-9 = C-terminal amide, Carba sulfide bridge: X-1 – Cys-6) | Deletion of Gln (−128.2 Da); Insertion of Asn (+114.1 Da) | Oxidation of Cys-11 to sulfoxide (+16 Da), sulfone (+32 Da), or sulfonate (+48 Da) | — |
| Leuprolide | XHWSYLLRP | Deletion of Ser-4 (−87.1 Da) or Arg-8 (−156.2 Da); Insertion of Trp (+186.2 Da), Arg (+156.2 Da), or His (+137.1 Da) | Oxidation to reduced Trp-3 (+2 Da) or hydroxy Trp-3 (+16 Da) | Substituted guanidine on Arg side chain (+126 Da) |
| Goserelin | XHWSYXLRP | Deletion of Pyr-1 (−111.1 Da) or Pro-8 (−97.1 Da); Insertion of Arg (+156.2 Da) or Tyr (+163.2 Da) | — | Arginine amination (+15 Da) |
| Triptorelin | XHWSYWLRPG | Deletion of Pyr-1 and Pro-9 (−111.1 Da + 97.1 Da); Insertion of His (+137.1 Da) or Tyr (+163.2 Da) | — | Arginine amination (+15 Da); Deamidation (+1.0 Da) |
| Calcitonin Salmon | CSNLSTCVLGKLSQELHKLQTYPRTNTGSGTP | Truncation at Lys and Arg residues | Oxidation at Cys-1/7 (+16 Da) | Arginine amination (+15 Da); Deamidation (+1.0 Da); Acetylation at cystine |
| Teduglutide | HGDGSFSDEMNTILDNLAARDFINWLIQTKITD | Deletion of Gly-4 (−57 Da); Insertion of Gly (+57 Da) | Oxidation at Met-10 (+16 Da) | Aspartic acid to Iso-aspartate isomerization |
| Teriparatide | SVSEIQLMHNLGKHLNSMERVEWLRKKLQDVHNF | Various truncation sequences | Oxidation at Met-8/18 (+16 Da) | Asp-10 acetate adduct (+70 Da); Asn to Asu modification (+57 Da); Succinimide formation (+97 Da) |
| Tirzepatide | YXEGTFTSDYSIXLDKIAQKAFVQWLIAGGPSSGAPPPS | Deletions at Tyr-10, Pro-Pro, Gly-4, Thr-5, and Gamma-Glu | — | L-isomer of Iso-Asp at residues 9 and 15; +4 Da kynurenine modification |
| Liraglutide | HAEGTFTSDVSSYLEGQAAKEEFIAWLVRGRG (gamma-E-palmitoyl at Glu-21) | Various truncation sequences; deletion of Ser-8 | Oxidation of Trp-31 (+16 Da) | Acetaldehyde adduct (+22.1 Da); D-isomer of Phe, Ser, and Thr; acetylation at Trp; +4 Da kynurenine at residue 25 |
| Semaglutide | HAEGTFTSDVSSYLEAQAAKEFQAWRNRGRG | Various truncation sequences; deletion of Thr-7; insertion of Gly at residues 29 or 31 | Oxidation of Trp-30 (+16 Da) | D-isomer of Phe, Ser, Ala, and Leu; +26 Da acetaldehyde Schiff base; +4 Da kynurenine at residue 25 |
Map Complex Agonists: Ensure complete confirmation of complex amino acid changes. Implement our validated GLP-1 Analog Peptide Sequencing Workflow for your modern synthetic target programs.
Establishing Precise Impurity Quantitation via Relative Response Factors
The Relative Response Factor (RRF) is used to compensate for differences in detector response between peptide impurities and the active pharmaceutical ingredient (API) during ultraviolet or mass spectrometric detection. Establishing reliable RRF values is essential for preventing both overestimation and underestimation of impurity concentrations during high-performance liquid chromatography (HPLC) analysis. Regulatory agencies, including the FDA and the European Medicines Agency (EMA), place significant value on RRF-corrected quantitation because it provides accurate mass balance calculations and dependable impurity profiling, even when isolated impurity reference standards are unavailable.
To determine the Relative Response Factor (RRF) for a specific peptide-related impurity, the Response Factor (RF), which represents the detector response generated per unit concentration, must first be established for both the active pharmaceutical ingredient (API) and the impurity. The response factors are defined as follows:
RFAPI = AAPI /CAPI and RFimpurity = Aimpurity /Cimpurity
where A denotes the integrated chromatographic peak area and C represents the known analyte concentration.
After determining these individual response factors, the Relative Response Factor (RRF) is calculated as the ratio of the impurity response factor to that of the API:
RRF = RFimpurity/ RFAPI
As an alternative approach, the RRF may be established using multi-point calibration curves by calculating the ratio of their respective calibration slopes:
RRF = Slopeimpurity /SlopeAPI
Once the RRF has been established, the true concentration of the impurity can be calculated by correcting for differences in detector sensitivity using the following equation:
Cimpurity = Aimpurity /RRF × RFAPI
The impurity concentration is then expressed as a percentage relative to the nominal concentration of the API according to the following relationship:
Impurity (%) = 100 × Cstd /CAPI × Aimpurity /Astd × 1 /RRF
where C₍std₎ and A₍std₎ represent the concentration and chromatographic peak area of the primary reference standard, respectively.
It is important to recognize that changes in HPLC instrumentation, mobile-phase composition, or column operating temperature may influence experimentally determined RRF values. Such variations can introduce quantitative uncertainty, particularly when the measured RRF falls outside the generally accepted range of 0.8 to 1.2. To eliminate the need for repeated synthesis and isolation of trace degradation products solely for RRF verification, many analytical laboratories employ an integrated quantitative Nuclear Magnetic Resonance–Liquid Chromatography (qNMR-LC) strategy.
Because the proton NMR integration signal is directly proportional to the molar quantity of an organic compound, regardless of its ultraviolet absorption characteristics, qNMR enables accurate determination of the molar ratio (n) of non-isolated impurities present within degraded peptide samples. By combining this experimentally determined molar ratio with molecular weight information obtained through LC-HRMS and the corresponding HPLC-UV peak areas, the Relative Response Factor can be directly calculated from the analytical mixture using the following equation:
RRF = Aimpurity /AAPI × MWimpurity /MWAPI × nAPI/ nimpurity
This integrated analytical strategy provides a scientifically rigorous and economically efficient approach for monitoring multiple unstable degradation products throughout every stage of a peptide product’s lifecycle.
De-risk Your Analytics: Acquire comprehensive quantitative data sets that stand up to rigorous regulatory checks. Partner with us for complete CRO for GLP-1 Peptide Characterization campaigns.
Conclusion: Strategic Value of Peptide Impurity Identification and Characterization Services
Comprehensive Peptide Impurity Identification and Characterization Services provide the high-resolution analytical data and regulatory documentation necessary to reduce development risks throughout peptide drug development programs. By implementing robust orthogonal analytical methodologies together with thoroughly validated stability-indicating studies, these services help ensure that peptide therapeutics satisfy global regulatory expectations for product safety, quality, and therapeutic equivalence.
Accurate characterization of both process-related and degradation-related impurities has become a fundamental requirement in modern biopharmaceutical development. Under the FDA guidance released in May 2021, developers of generic synthetic peptides are required to structurally identify and qualify every peptide-related impurity present at concentrations of at least 0.10%. Meeting this regulatory expectation is particularly demanding because peptide impurities frequently arise from stereoisomeric conversions, sequence-dependent degradation pathways, and structurally similar by-products. Successfully addressing these analytical challenges requires moving beyond conventional one-dimensional chromatographic methods and adopting advanced orthogonal analytical platforms capable of delivering superior structural resolution.
Partnering with an organization specializing in comprehensive Peptide Impurity Identification and Characterization Services enables pharmaceutical and biotechnology companies to streamline development programs, successfully navigate regulatory inspections, and strengthen patient safety initiatives. By integrating advanced high-resolution mass spectrometry (HRMS), two-dimensional liquid chromatography (2D-LC), and qNMR-based Relative Response Factor determination, these services generate regulatory-ready analytical data packages that support the successful development and commercialization of highly purified peptide therapeutics.
Take the Next Step: Ready to accelerate your primary structural validations? Reach out to discover our specialized GLP-1 Peptide Sequencing CRO Services and secure regulatory-ready data packs today.
To learn more about optimizing analytical workflows, performing stability-indicating method validation, or initiating a peptide sameness study, please contact the Ph.D.-level scientists at the ResolveMass Laboratories Contact Page.
Frequently Asked Questions
For generic synthetic peptide products submitted through an ANDA, the FDA requires any peptide-related impurity present at 0.10% or higher to be structurally identified and thoroughly characterized. In addition, any newly identified specified impurity generally should not exceed 0.5% unless it is scientifically justified. The European Pharmacopoeia (Ph. Eur.) Monograph 2034 follows a different approach by setting an identification threshold of 0.5% and a qualification limit of 1.0%, making the FDA requirements comparatively more stringent.
Residual acetic acid in Fmoc-amino acid starting materials acts as an irreversible chain-terminating reagent during peptide synthesis. It reacts with the free amino group of the growing peptide chain, preventing the addition of subsequent amino acid residues. Even trace quantities can significantly affect synthesis because the low molecular weight of acetic acid results in a relatively high molar concentration. Consequently, small amounts of contamination may terminate a substantial percentage of peptide chains and reduce the overall manufacturing yield.
Base-catalyzed aspartimide formation commonly occurs during Fmoc deprotection when secondary amines, such as piperidine, remove a proton from the backbone amide nitrogen adjacent to an Asp residue. This promotes an intramolecular nucleophilic attack on the side-chain β-carboxyl group, producing a cyclic succinimide intermediate. The intermediate is chemically unstable and subsequently undergoes hydrolysis, generating both the native α-aspartyl peptide and the isomerized β-aspartyl (isoaspartyl) form. This pathway is one of the major sources of peptide degradation and sequence heterogeneity.
The mechanism of deamidation depends largely on the pH of the surrounding environment. Under acidic conditions (pH below 3), the reaction proceeds primarily through direct hydrolysis of the asparagine side-chain amide, producing the native α-aspartyl peptide. At neutral or alkaline pH, the process involves formation of a cyclic imide intermediate before hydrolysis occurs. As a result, both α-aspartyl and isoaspartyl peptides are generated, increasing structural heterogeneity within the peptide sample.
Leucine and Isoleucine possess identical molecular masses, making them difficult to distinguish using conventional MS/MS analysis alone. Advanced mass spectrometric techniques employ low-energy collision-induced dissociation followed by MS³ analysis of the m/z 86 immonium ion. This additional fragmentation step produces characteristic product ions at m/z 30, 44, and 69, whose relative intensities provide a reliable means of differentiating between these two isobaric amino acids.
Quantitative Nuclear Magnetic Resonance (qNMR) provides an accurate and detector-independent method for determining Relative Response Factors (RRFs). Since proton NMR signal intensity is directly proportional to molar concentration, it enables precise quantification without relying on ultraviolet absorbance properties. By comparing NMR-derived molar ratios with HPLC-UV peak areas, laboratories can calculate reliable RRF values even when purified impurity reference standards are unavailable. This approach improves quantitative accuracy while reducing analytical costs.
Two-dimensional liquid chromatography (2D-LC) addresses mobile phase incompatibility by using an automated heart-cutting interface to isolate selected impurity peaks from the first chromatographic dimension. These fractions are subsequently desalted online and transferred into a volatile mobile phase that is fully compatible with mass spectrometric detection. This workflow preserves chromatographic resolution while allowing direct coupling between pharmacopoeial HPLC methods and high-resolution LC-MS analysis. As a result, both sensitivity and structural characterization are significantly improved.
Disulfide scrambling alters the native disulfide bond arrangement within therapeutic peptides, leading to changes in their three-dimensional structure. These conformational alterations may expose previously hidden hydrophobic regions or generate new antigenic epitopes that were not present in the correctly folded molecule. Such structural modifications can activate both innate and adaptive immune responses, increasing the likelihood of anti-drug antibody formation and potentially reducing therapeutic efficacy.
Demonstrating API sameness requires a comprehensive comparison between multiple batches of the generic synthetic peptide and the corresponding reference listed drug (RLD). These studies typically evaluate the primary amino acid sequence, physicochemical properties, secondary and tertiary structures, aggregation behavior, biological activity, and impurity profile. Regulatory agencies generally expect comparative data from several manufacturing batches to ensure consistency and establish therapeutic equivalence. Comprehensive analytical characterization provides the scientific evidence necessary to support regulatory approval.
Reference:
- Malonis, R. J., Lai, J. R., & Vergnolle, N. (2014). Peptide-based vaccines: Current progress and future challenges. Chemical Reviews, 114(18), 978–1004. https://doi.org/10.1021/cr400694p
- Kuril, A. K. (2025). The critical need for implementing RRF in the accurate assessment of impurities in peptide therapeutics. Analytical Chemistry, 97(24), 12480–12485. https://doi.org/10.1021/acs.analchem.5c02149
- U.S. Food and Drug Administration. (2021, May). ANDAs for certain highly purified synthetic peptide drug products that refer to listed drugs of recombinant DNA (rDNA) origin: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/166572/download
- U.S. Food and Drug Administration. (2021, May). ANDAs for certain highly purified synthetic peptide drug products that refer to listed drugs of recombinant DNA (rDNA) origin: Guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/andas-certain-highly-purified-synthetic-peptide-drug-products-refer-listed-drugs-rdna-origin
- Duncan, K. (2024, April 9–10). CMC regulatory experiences and expectations for peptides [Conference presentation]. USP Workshop on Therapeutic Peptides and Oligonucleotides: Regulations and Quality Standards, United States Pharmacopeia. https://www.usp.org/sites/default/files/usp/document/events-and-training/03-CMC-Regulatory-Experiences-and-Expectations_Katharine-Duncan.pdf
- Bosc-Bierne, G., & Weller, M. G. (2025). Investigation of impurities in peptide pools. Separations, 12(2), 36. https://doi.org/10.3390/separations12020036

