
Introduction to Peptide-Oligonucleotide Conjugates Specification Setting
Peptide-Oligonucleotide Conjugates Specification Setting is the systematic process of establishing scientifically justified physical, chemical, and biological acceptance criteria to ensure the quality, safety, and therapeutic performance of chimeric biotherapeutics. These specifications form the foundation of an effective quality control strategy by defining the requirements that a drug substance must satisfy before it can be released for clinical use. Achieving this objective requires advanced analytical methodologies capable of simultaneously evaluating the polypeptide carrier, the oligonucleotide payload, and the covalent linker that joins these two functional components.
Learn more about QC testing for peptide-oligonucleotide conjugates and how comprehensive analytical testing supports reliable specification setting.
Developing a reliable control strategy for peptide-oligonucleotide conjugates requires analytical scientists to address the significant physicochemical differences that naturally exist within these hybrid molecules. Oligonucleotides possess highly hydrophilic, polyanionic phosphodiester or phosphorothioate backbones, whereas peptide components often contain hydrophobic regions or highly basic amino acid sequences, such as arginine-rich cell-penetrating peptides. The coexistence of these contrasting structural features increases the likelihood of self-aggregation, electrostatic interactions, and multiple forms of chemical degradation throughout development and storage.
Explore the major challenges in peptide-oligonucleotide conjugates that influence analytical development and quality control.
Because of these unique molecular characteristics, conventional analytical workflows designed for small molecules or monoclonal antibodies are generally inadequate for evaluating peptide-oligonucleotide conjugates. Instead, organizations must rely on specialized bioconjugate contract research laboratories that can develop phase-appropriate analytical specifications tailored to these complex therapeutic platforms.
ResolveMass Laboratories Inc., an ISO 9001:2015-certified and USFDA-registered facility (FDA Establishment Identifier: 3042696771), utilizes advanced high-resolution mass spectrometry (HRMS), quantitative nuclear magnetic resonance (qNMR), and multidimensional liquid chromatography to overcome the analytical challenges associated with comprehensive bioconjugate characterization.
Discover ResolveMass’ expertise in structural characterization of peptide-oligonucleotide conjugates using advanced analytical technologies.
Article Summary:
- Peptide-oligonucleotide conjugate (POC) specification setting establishes scientifically justified quality standards to ensure the safety, efficacy, stability, and consistency of these hybrid therapeutics using advanced analytical techniques.
- Critical quality attributes (CQAs) such as the peptide-to-oligonucleotide ratio, linker stability, aggregation, stereochemical purity, and structural integrity must be tightly controlled, as they directly impact therapeutic performance and patient safety.
- Regulatory compliance requires a hybrid approach that combines guidance for peptides and oligonucleotides, with strict monitoring of sequence-related, process-related, and degradation impurities to meet evolving global quality standards.
- Advanced analytical technologies, including high-resolution mass spectrometry, multidimensional chromatography, and orthogonal purification methods, are essential for confirming molecular identity, conjugation sites, purity, and impurity profiles of these complex bioconjugates.
- Comprehensive stability and toxicology assessments evaluate degradation pathways, pharmacokinetics, tissue-specific behavior, aggregation, and conjugate-specific toxicity to ensure long-term product stability and safe clinical performance.
- Phase-appropriate GMP release specifications define acceptance criteria for identity, purity, conjugation efficiency, residual contaminants, microbiological quality, and other critical parameters, becoming increasingly stringent as products progress from clinical development to commercialization.
- A robust specification strategy integrating advanced analytical characterization, regulatory compliance, and lifecycle quality control minimizes development risks, supports successful regulatory submissions, and accelerates the development of next-generation peptide-oligonucleotide therapeutics.
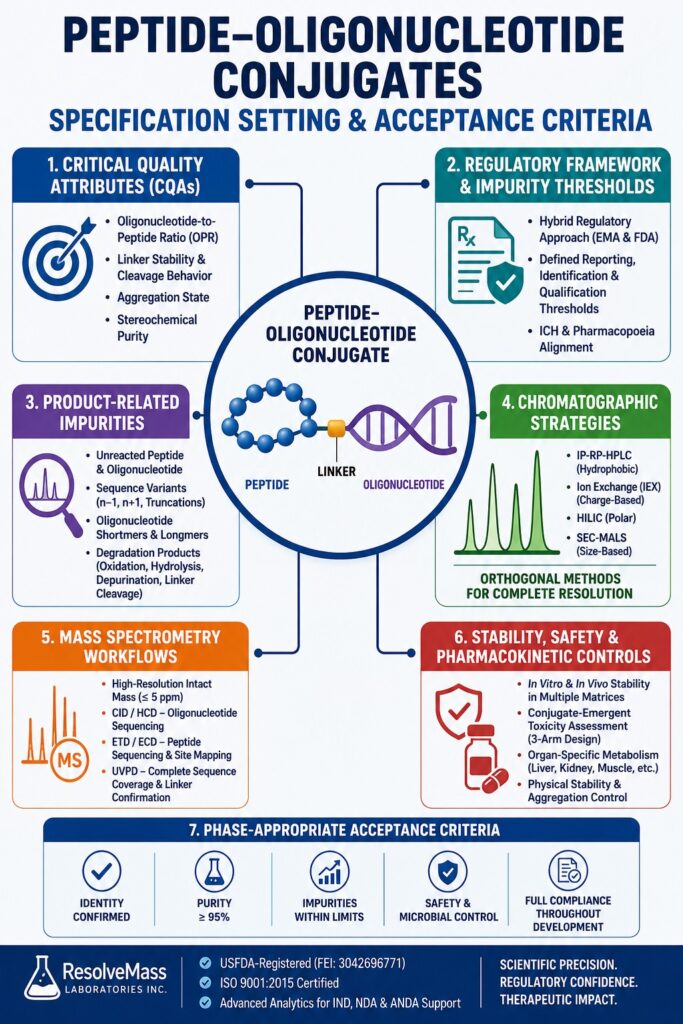
Establishing Critical Quality Attributes for Peptide-Oligonucleotide Conjugates Specification Setting
Defining critical quality attributes (CQAs) for Peptide-Oligonucleotide Conjugates Specification Setting involves identifying the physical, chemical, and biological characteristics that directly influence therapeutic safety, long-term stability, and biological activity. Since these conjugates combine two chemically distinct molecular domains, their CQAs are inherently multidimensional and require comprehensive analytical evaluation.
One of the most important CQAs is the oligonucleotide-to-peptide ratio (OPR). Maintaining strict control of this ratio is essential because any deviation may reduce biological activity or increase the potential for unintended off-target effects. During chemical synthesis, incorrect reaction stoichiometry or incomplete conjugation can generate mixtures containing unreacted starting materials, intended 1:1 conjugates, and undesired multi-conjugated species, including 1:2 and 1:3 variants.
Another essential quality attribute is the stability and cleavage behavior of the linker chemistry. Whether the conjugate employs a stable thioether linkage or a cleavable disulfide or enzyme-responsive linker, its cleavage kinetics must be thoroughly characterized. Premature linker cleavage can compromise targeted delivery, reduce therapeutic effectiveness, and significantly alter pharmacokinetic behavior. Additional physical quality attributes, including aggregation state and stereochemical purity, must also be carefully monitored because self-assembled aggregates may increase immunogenicity and influence cell-penetrating efficiency.
Learn how peptide-oligonucleotide conjugate linker chemistry influences stability, release profiles, and therapeutic performance.
Regulatory Frameworks and Impurity Thresholds for Bioconjugate Specifications
Regulatory expectations for impurity limits in bioconjugate specifications are derived from the combined principles governing small molecules, synthetic peptides, and synthetic oligonucleotides. Since peptide-oligonucleotide conjugates (POCs) do not fit neatly within existing regulatory classifications, health authorities generally apply a hybrid regulatory framework that integrates multiple guidance documents.
Historically, synthetic peptides and synthetic oligonucleotides were either fully or partially excluded from the scope of International Council for Harmonisation (ICH) guidelines Q3A/B, Q6A/B, and M7. However, the European Medicines Agency (EMA) introduced a synthetic peptide quality guideline, adopted in December 2025 and effective from June 1, 2026, together with a draft guideline for synthetic oligonucleotides that establishes harmonized impurity control strategies.
The EMA framework is aligned with the general monograph of the European Pharmacopoeia entitled Substances for Pharmaceutical Use, providing clearly defined impurity limits based on molecular complexity. For synthetic oligonucleotides, the draft guidance proposes harmonized reporting, identification, and qualification thresholds that reflect the capabilities of modern chromatographic technologies.
| Impurity Parameter | Synthetic Peptides (EMA 2026) | Synthetic Oligonucleotides (Draft EMA) | FDA Generic Peptide Guidelines |
|---|---|---|---|
| Reporting Threshold | ≥ 0.10% (or 0.1%) | ≥ 0.20% (or 0.2%) | ≥ 0.10% (or 0.1%) |
| Identification Threshold | ≥ 0.50% [cite: 6] | ≥ 1.0% [cite: 5, 7, 30] | ≥ 0.10% (or 0.1%) |
| Qualification Threshold | ≥ 1.0% [cite: 6] | ≥ 1.5% [cite: 5, 7, 30] | > 0.50% for newly specified impurities |
| Purity Basis Specification | Counter-ion free, anhydrous | Sodium salt form, anhydrous | Comparable impurity profile to the Reference Listed Drug (RLD) |
These regional regulatory recommendations provide valuable guidance when establishing scientifically justified acceptance criteria. Although the U.S. Food and Drug Administration (FDA) has not formally adopted the EMA impurity limits for synthetic therapeutics, its nonclinical safety guidance and abbreviated new drug application (ANDA) requirements follow comparable impurity control principles. Both agencies emphasize rigorous identification of sequence-related impurities, stereoisomeric variants, and process-related genotoxic contaminants.
Characterizing Product-Related Impurities in Peptide-Oligonucleotide Conjugates Specification Setting
Product-related impurities in Peptide-Oligonucleotide Conjugates Specification Setting are molecular variants generated during synthesis, conjugation, purification, or storage that remain structurally related to the intended therapeutic molecule. These impurities require careful identification and strict control because they may compete with the target molecule for receptor binding, reduce therapeutic efficacy, or provoke undesirable immune responses.
During the conjugation process, incomplete reaction conversion or poor stoichiometric control frequently results in residual unconjugated peptide, free oligonucleotide, and multiple conjugated species within the reaction mixture. These process-related impurities must be maintained within well-defined specification limits to ensure consistent product quality.
Sequence-related impurities originating during solid-phase synthesis must also be comprehensively characterized. Within the peptide domain, these impurities include amino acid deletion sequences (n−1), amino acid insertion sequences (n+1), truncated peptide fragments, and diastereomeric products arising from amino acid racemization.
Similarly, the oligonucleotide component may contain characteristic impurities such as n−x shortmer failure sequences resulting from incomplete coupling and capping reactions, n+x longmer species, depurinated oligonucleotides, and diastereomeric variants.
During storage, multiple degradation pathways can generate additional related substances requiring stability-indicating specifications. These degradation mechanisms include peptide oxidation, particularly at methionine and cysteine residues, deamidation of asparagine or glutamine residues, peptide hydrolysis, oligonucleotide depurination, and linker cleavage, all of which may significantly affect therapeutic performance.
Learn more about peptide-oligonucleotide conjugate stability and the analytical approaches used to monitor degradation pathways throughout development and storage.
Advanced Purification and Chromatography in Peptide-Oligonucleotide Conjugates Specification Setting
Effective chromatographic separation of peptide-oligonucleotide conjugates requires the use of orthogonal separation mechanisms, wide-pore stationary phases, and carefully optimized operating conditions to achieve baseline resolution. Owing to the combined ionic and hydrophobic characteristics of these hybrid molecules, single-mode chromatographic approaches rarely provide sufficient separation efficiency.
Developing a robust purification and analytical workflow requires integrating multiple complementary chromatographic techniques capable of resolving the highly complex mixtures produced during bioconjugate synthesis. The principal purification strategies are summarized below.
| Purification Mode | Primary Chromatographic Principle | Target Impurities and Analytes | Mobile Phase Configuration |
|---|---|---|---|
| IP-RP-HPLC [cite: 3] | Hydrophobic retention enhanced by ion-pairing reagents on reversed-phase media | Unreacted modified or unmodified oligonucleotides, linker precursors, truncated peptide sequences | Phase A: 0.1 M Triethylammonium Acetate (TEAA) or Hexafluoroisopropanol (HFIP) with Triethylamine (TEA); Phase B: 100% Acetonitrile (ACN) |
| Ion Exchange (IEX) [cite: 3] | Electrostatic separation based on charge differences | n−1 deletion sequences, shortmers, truncated species, and multi-conjugated variants | Phase A: Aqueous Tris-HCl buffer (pH 7.0–8.0); Phase B: Tris-HCl buffer containing 1.0–1.5 M Sodium Chloride |
| HILIC [cite: 3] | Polar partitioning between a water-rich stationary layer and an organic-rich mobile phase | Highly polar degradation products, residual uncoupled linkers, and hydrophilic peptide impurities | Phase A: Aqueous Ammonium Acetate buffer (pH 5.0–6.0); Phase B: 95% Acetonitrile in water |
| SEC-MALS [cite: 3, 21] | Size-exclusion based on hydrodynamic volume | High-molecular-weight aggregates, self-assembled complexes, and polymeric degradation products | Phosphate-Buffered Saline (PBS) containing elevated salt concentrations to minimize non-specific ionic interactions |
Successful implementation of these chromatographic techniques requires optimized column selection and carefully controlled operating conditions. To minimize metal-induced sample adsorption and reduce peak tailing associated with phosphate-containing oligonucleotide backbones, wide-pore bioinert columns, such as YMC Accura Triart Bio C18 columns with 300 Å pore diameters, should be employed.
Elevated column temperatures ranging from 60°C to 80°C are also recommended because they help disrupt secondary DNA or RNA structures and peptide folding, thereby reducing mass transfer resistance and improving chromatographic resolution. Furthermore, mobile phase pH should generally be maintained between pH 6.0 and 8.0, since acidic conditions below pH 6.0 reduce separation efficiency, whereas alkaline conditions above pH 8.0 accelerate stationary-phase degradation.
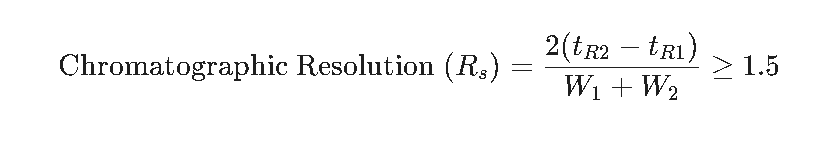
Explore proven peptide-oligonucleotide conjugate synthesis methods that support efficient purification and high-quality bioconjugate production.
Mass Spectrometry Workflows for Structural Verification and Linker Site Mapping
Verification of intact molecular weight and precise localization of conjugation sites is primarily accomplished using high-resolution mass spectrometry (HRMS) in combination with multiple tandem mass spectrometric fragmentation techniques. Given the inherent structural heterogeneity of peptide-oligonucleotide conjugates, accurate mass determination serves as one of the most reliable indicators of structural integrity.
For intact mass analysis, electrospray ionization (ESI) coupled with Orbitrap or Q-TOF mass spectrometers remains the preferred analytical platform because it provides exceptional mass accuracy and high resolving power within 5 ppm. Since ESI is particularly susceptible to sodium and potassium adduct formation, volatile ion-pairing reagents are typically employed during liquid chromatography to minimize salt-related interference.
Mass Measurement Error (ppm) = [(Observed Mass − Theoretical Mass) / Theoretical Mass] × 10⁶ ≤ 5 ppm
Alternatively, matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry provides a valuable option for rapid, high-throughput analysis of crude reaction mixtures. MALDI-TOF exhibits greater tolerance toward salts and buffer components than ESI, although its overall mass accuracy and resolution are generally lower.
To establish sequence identity and accurately localize the covalent conjugation site, analysts employ complementary tandem mass spectrometry (MS/MS) fragmentation strategies.
Collision-Induced Dissociation (CID) and Higher-Energy Collisional Dissociation (HCD)
CID and HCD predominantly transfer fragmentation energy to the phosphodiester or phosphorothioate backbone of the oligonucleotide component. These fragmentation pathways generate characteristic a, w, c, and d ion series that enable accurate sequence verification of the nucleic acid segment while largely preserving the integrity of the peptide domain.
Electron Transfer Dissociation (ETD) and Electron Capture Dissociation (ECD)
ETD and ECD selectively cleave the peptide amide backbone, producing characteristic c and z ion series while preserving labile post-translational modifications, sugar modifications, and the covalent linker. These fragmentation mechanisms facilitate precise peptide sequencing and accurately identify the amino acid residue involved in conjugation.
Ultraviolet Photodissociation (UVPD)
Ultraviolet Photodissociation (UVPD) utilizes high-energy ultraviolet photons, typically at wavelengths of 193 nm or 213 nm, to simultaneously fragment both the peptide and oligonucleotide domains during a single mass spectrometric experiment. Unlike conventional fragmentation techniques that preferentially target one molecular component, UVPD produces extensive fragmentation across the entire bioconjugate. This comprehensive fragmentation pattern enables complete sequence coverage of the intact peptide-oligonucleotide conjugate while simultaneously confirming the precise chemical bond at the conjugation site, making it a powerful tool for structural verification.
Learn more about CMC services for peptide-oligonucleotide conjugates and the advanced analytical workflows used for structural verification and quality assessment.
Toxicology, Stability, and Pharmacokinetic Controls
Establishing scientifically sound specifications for peptide-oligonucleotide bioconjugates requires comprehensive evaluation of both in vitro and in vivo stability across multiple biological matrices to accurately characterize degradation pathways. Because these hybrid therapeutics combine two distinct pharmacophores within a single molecular entity, their pharmacokinetic behavior, metabolism, and toxicological profiles are considerably more complex than those of conventional therapeutic agents.
One of the primary concerns during bioconjugate development is conjugate-emergent toxicity. This phenomenon occurs when the intact peptide-oligonucleotide conjugate demonstrates toxicological effects that are absent in either the unconjugated peptide or the free oligonucleotide alone. To adequately assess this risk, preclinical toxicology programs should incorporate a three-arm experimental design that independently evaluates the free peptide, the free oligonucleotide, and the intact conjugate under identical experimental conditions. This comparative approach enables investigators to identify toxicities that arise specifically from the conjugated molecular architecture.
In addition to systemic toxicology, tissue-specific metabolic behavior must be investigated using matrix-specific stability studies performed in homogenates prepared from relevant target organs, including the liver and kidneys. For oligonucleotide therapeutics designed to target extrahepatic tissues, such as kidney tubular epithelial cells or skeletal muscle, evaluating conjugate stability in the presence of intracellular proteases and nucleases becomes an essential quality attribute. These studies help establish whether the conjugate remains sufficiently stable to achieve targeted delivery while maintaining its intended biological activity.
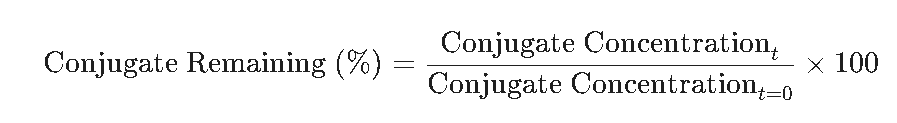
Discover how preclinical services for peptide-oligonucleotide conjugates support toxicology assessments, stability studies, and early-stage development.
Furthermore, physical stability specifications must be established to minimize the risk of self-association and aggregation throughout storage and distribution. Lyophilization is widely employed to preserve long-term stability; however, both freezing and drying stages may introduce physical stresses capable of inducing peptide denaturation or promoting aggregate formation. Consequently, product specifications should require the use of well-characterized cryoprotectants such as sucrose or trehalose, suitable bulking agents, and carefully selected nonionic surfactants. Physical aggregation should be routinely monitored using size-exclusion chromatography with multi-angle light scattering (SEC-MALS) together with dynamic light scattering (DLS) to ensure consistent product quality during storage.
Learn more about peptide-oligonucleotide conjugates pharmacokinetics and the factors influencing in vivo stability and therapeutic performance.
Establishing Acceptance Criteria for Phase-Appropriate Compliance
A comprehensive release specification for GMP-grade peptide-oligonucleotide conjugates should encompass physical appearance, structural identity, chemical purity, process-related impurities, residual contaminants, and microbiological quality attributes. These specifications must remain phase-appropriate throughout product development, allowing relatively flexible acceptance criteria during early clinical investigations while gradually transitioning to tightly controlled, fully validated commercial release specifications as regulatory expectations increase.
A representative release specification matrix for a GMP-grade peptide-oligonucleotide conjugate drug substance is presented below.
| Test Parameter | Analytical Methodology | Acceptance Criteria (GMP Release) | Scientific Justification |
|---|---|---|---|
| Appearance | Visual inspection | White to off-white lyophilized cake or powder | Confirms successful lyophilization and acceptable physical appearance. |
| Intact Mass Identity | High-Resolution Orbitrap or Q-TOF ESI-MS | Matches the calculated theoretical mass (±5 ppm) | Verifies the correct elemental composition of the bioconjugate. |
| Sequence Identity | LC-MS/MS mapping following enzymatic digestion | Matches the theoretical peptide and oligonucleotide sequences | Confirms primary structure while eliminating concerns regarding synthesis or conjugation errors. |
| Conjugation Stoichiometry (OPR) | Native SEC-MS or high-resolution IP-RP-HPLC | 1:1 conjugate species ≥98.0%; multi-conjugated species ≤1.0% | Ensures the intended ligand-to-payload ratio for optimal therapeutic performance. |
| Purity (Conjugate Peak) | Orthogonal IP-RP-HPLC and Strong Anion Exchange (IEX) | ≥95.0% of the total chromatographic peak area | Demonstrates high product purity and manufacturing consistency. |
| Unreacted Peptide | Reversed-Phase HPLC | ≤1.0% [cite: 9] | Limits exposure to unconjugated targeting or cell-penetrating peptides. |
| Unreacted Oligonucleotide | Strong Anion Exchange HPLC | ≤1.0% [cite: 9] | Restricts the presence of free, non-targeted oligonucleotide payloads. |
| Related Substances | High-resolution IP-RP-LC-MS | Specified impurities ≤1.0%; unspecified impurities ≤0.20%; total impurities ≤5.0% | Controls synthesis-related failure sequences and degradation products. |
| Residual TFA Content | High-Performance Anion Exchange Chromatography (HPAEC) with Conductivity Detection | ≤0.1% (w/w) | Monitors residual trifluoroacetic acid used during peptide synthesis. |
| Sodium (Counter-ion) Content | Inductively Coupled Plasma Mass Spectrometry (ICP-MS) | Report actual results; typical target range 5.0%–10.0% | Confirms a consistent sodium salt form of the oligonucleotide backbone. |
| Residual Solvents | Headspace Gas Chromatography (GC-FID or GC-MS) | Complies with ICH Q3C limits for Acetonitrile, DMF, and Pyridine | Ensures adequate removal of organic synthesis and cleavage solvents. |
| Elemental Impurities | ICP-MS | Copper ≤15 ppm (when CuAAC chemistry is used); heavy metals within ICH Q3D limits | Limits residual catalytic metals originating from the conjugation process. |
| Moisture Content (Water) | Coulometric Karl Fischer Titration | ≤10.0% (or ≤15.0%) | Prevents hydrolytic degradation during storage. |
| Bacterial Endotoxins | Limulus Amebocyte Lysate (LAL) Test (USP <85>) | ≤5.0 EU/kg body weight | Critical safety requirement for parenteral pharmaceutical products. |
| Bioburden | Aerobic Count and Spore Count (USP <61>/<62>) | ≤10 CFU/g | Confirms microbiological quality and manufacturing control of the drug substance. |
Explore comprehensive CMC services for peptide-oligonucleotide conjugates to support GMP release testing, analytical validation, and quality compliance.
Conclusion: Future-Proofing Peptide-Oligonucleotide Conjugates Specification Setting
Implementing a comprehensive and scientifically rigorous approach to Peptide-Oligonucleotide Conjugates Specification Setting is essential for minimizing development risks, supporting successful clinical advancement, and achieving regulatory approval for next-generation genetic medicines. Because these hybrid therapeutics possess the biological characteristics of both peptides and oligonucleotides, their specification strategies must incorporate advanced multi-attribute analytical methodologies capable of evaluating every critical quality attribute throughout the product lifecycle.
The integration of orthogonal chromatographic separation techniques, wide-pore bioinert stationary phases, and high-resolution mass spectrometric fragmentation enables analytical scientists to accurately identify synthesis-related sequence variants, degradation products, linker modifications, and process-related impurities with exceptional confidence. These comprehensive analytical strategies strengthen product characterization while supporting robust quality control and regulatory compliance.
ResolveMass Laboratories Inc. supports pharmaceutical and biotechnology organizations throughout every stage of drug development, from early research and development through validated commercial drug substance submissions. Through its USFDA-registered facility, ISO 9001:2015-certified quality management system, and extensive expertise in high-resolution mass spectrometry, multidimensional chromatography, and biopolymer characterization, ResolveMass delivers the analytical validation necessary to support IND, NDA, and ANDA regulatory submissions.
Learn how ResolveMass supports peptide-oligonucleotide conjugates in IND submissions with analytical characterization, regulatory documentation, and CMC expertise.
Organizations seeking clinical-phase analytical validation or customized peptide-oligonucleotide conjugate characterization programs are encouraged to contact the technical specialists at ResolveMass Laboratories Inc. through the ResolveMass Contact Us page to discuss project-specific analytical requirements.
Frequently Asked Questions
A risk-based, science-driven approach should be used when defining impurity thresholds for peptide-oligonucleotide conjugates. Since these therapeutics combine peptide and oligonucleotide components, specifications should incorporate principles from both synthetic peptide regulations and emerging oligonucleotide guidance. In general, impurities above 0.20% should be reported, those reaching 1.0% should undergo structural identification, and impurities at or above 1.5% require toxicological qualification or appropriate scientific justification before regulatory acceptance.
Each phosphorothioate linkage introduces a new chiral phosphorus center, creating numerous stereoisomeric forms within a single oligonucleotide molecule. As the number of phosphorothioate linkages increases, the possible stereochemical combinations expand dramatically, making complete chromatographic separation impractical. For this reason, analytical specifications generally emphasize consistent stereochemical profiles between manufacturing batches using complementary techniques such as 31P NMR, circular dichroism (CD), and validated ion-pair reversed-phase HPLC rather than attempting to isolate every individual diastereomer.
Residual unconjugated peptide and free oligonucleotide can significantly influence the safety, efficacy, and pharmacokinetic profile of the finished bioconjugate. Free peptides may bind to target receptors without delivering the therapeutic oligonucleotide, while unconjugated oligonucleotides are more susceptible to rapid renal elimination and may increase kidney exposure. To maintain consistent product quality, release specifications typically restrict both impurities to concentrations of no more than 1.0%.
Large peptide-oligonucleotide conjugates require chromatographic columns with wide pore diameters to ensure efficient molecular diffusion and accurate separation. Columns with approximately 300 Å pore sizes provide improved recovery and sharper peak shapes compared with conventional 100 Å materials. In addition, bioinert hardware, including polymer-coated or ceramic-lined flow paths, minimizes unwanted interactions between metal surfaces and the negatively charged oligonucleotide backbone, thereby improving reproducibility and reducing peak tailing.
Characterizing linker stability requires analytical methods capable of monitoring conjugate integrity under biologically relevant conditions. Cleavable linkers are commonly evaluated by exposing the conjugate to reducing agents or specific enzymes and measuring the rate of bond cleavage over time. Orthogonal techniques such as ion-pair reversed-phase HPLC, capillary gel electrophoresis (CGE), and LC-MS provide detailed information about linker degradation, fragment formation, and overall release kinetics.
The 2026 EMA guideline places greater emphasis on comprehensive characterization and control of peptide starting materials throughout the manufacturing process. Manufacturers are expected to justify the selection of amino acid building blocks, resin supports, and intermediate fragments while demonstrating appropriate chemical and stereochemical purity. In addition, impurity fate and purging studies should confirm that contaminants originating from starting materials are effectively removed and do not contribute to impurities in the final conjugated drug substance.
A three-arm toxicology study separately evaluates the free peptide, the free oligonucleotide, and the intact peptide-oligonucleotide conjugate under comparable experimental conditions. This strategy helps distinguish toxicities that arise exclusively from the conjugated molecule rather than from its individual components. Such studies also provide valuable insight into tissue distribution, metabolic behavior, immunogenicity, and any unexpected safety concerns associated with the complete bioconjugate.
Inductively Coupled Plasma Mass Spectrometry (ICP-MS) is routinely used to quantify trace metal contaminants remaining after copper-catalyzed azide-alkyne cycloaddition (CuAAC) reactions. Because residual copper can promote oxidation and contribute to product toxicity, highly sensitive elemental analysis is required before product release. ICP-MS offers parts-per-billion detection capability, enabling manufacturers to verify that purification processes have reduced copper concentrations to acceptable regulatory limits, typically not exceeding 15 ppm.
Unlike recombinant biologics, synthetic peptides and oligonucleotides possess well-defined chemical structures that can be thoroughly characterized using advanced analytical technologies. High-resolution mass spectrometry, sequence-specific LC-MS/MS, and orthogonal chromatographic techniques provide detailed confirmation of molecular identity, purity, and sequence integrity. When these analytical methods sufficiently demonstrate product quality, regulatory authorities generally accept the omission of routine biological potency assays from release testing.
Although lyophilization greatly improves long-term product stability, freezing and dehydration can expose peptide domains to physical stress, increasing the possibility of unfolding and aggregate formation. Appropriate formulation strategies, including the use of cryoprotectants and stabilizing excipients, help minimize these effects. Product specifications should establish strict limits for high-molecular-weight aggregates, commonly not exceeding 1.0% as measured by SEC-MALS, while also confirming acceptable cake appearance and complete reconstitution within approximately five minutes.
Reference:
- Al Bardawil, S., Barthélémy, P., & Ferey, L. (2026). Advances in analysis of therapeutic oligonucleotides with chromatography coupled to mass spectrometry. Analytical Chemistry, 98(15), 10895–10912. https://doi.org/10.1021/acs.analchem.5c06656
- Thürmer, R. (2024, April 9). European quality guidelines for synthetic peptides and oligonucleotides [Presentation]. USP Workshop on Peptide and Oligonucleotide Therapeutics, Rockville, MD. United States Pharmacopeia. https://www.usp.org/sites/default/files/usp/document/our-work/biologics/documents/Rene%20Thurmer_European%20quality%20guidelines.pdf
- Elsayed, Y. Y., Kühl, T., & Imhof, D. (2025). Regulatory guidelines for the analysis of therapeutic peptides and proteins. Journal of Peptide Science, 31(3), e70001. https://doi.org/10.1002/psc.70001
- Naganuma, M., Tsuji, G., Amiya, M., Hirai, R., Higuchi, Y., Hata, N., Nozawa, S., Watanabe, D., Nakajima, T., & Demizu, Y. (2025). High-resolution HPLC for separating peptide–oligonucleotide conjugates. ACS Omega. Advance online publication. https://doi.org/10.1021/acsomega.5c01308
- European Medicines Agency. (2022, September 15). Concept paper on the establishment of a guideline on the development and manufacture of synthetic oligonucleotides (EMA/CHMP/QWP/735423/2022). https://www.ema.europa.eu/en/documents/scientific-guideline/concept-paper-establishment-guideline-development-and-manufacture-synthetic-oligonucleotides_en.pdf
- U.S. Food and Drug Administration. (2021, May). ANDAs for certain highly purified synthetic peptide drug products that refer to listed drugs of rDNA origin: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/107622/download
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2006). ICH harmonised tripartite guideline Q3A(R2): Impurities in new drug substances. https://database.ich.org/sites/default/files/Q3A%28R2%29%20Guideline.pdf

