
Introduction:
PLGA Formulation Development Services encompass a highly specialized range of contract research capabilities aimed at transforming complex active pharmaceutical ingredients (APIs) into precisely engineered, long-acting parenteral depot formulations. These services facilitate controlled modulation of drug release profiles over periods extending from several weeks to multiple months by utilizing the predictable hydrolytic degradation characteristics of poly(lactic-co-glycolic acid) (PLGA) copolymers. The therapeutic value of these biodegradable polymer systems lies in their ability to sustain consistent plasma drug concentrations, eliminate first-pass metabolism, and significantly enhance patient adherence by converting daily oral therapies into monthly or extended-duration injectable treatments. Successfully developing advanced long-acting injectable (LAI) formulations requires an integrated understanding of polymer science, sophisticated microencapsulation technologies, and comprehensive analytical characterization. Through structured Quality by Design (QbD) methodologies, contract research organizations (CROs) systematically evaluate critical material attributes (CMAs) and critical process parameters (CPPs) to ensure batch-to-batch consistency while meeting rigorous regulatory bioequivalence requirements.
Need expert support for your project? Explore our PLGA long-acting injectable formulation services.
Share via:
Article Summary:
- PLGA formulation development services enable the creation of long-acting injectable drug products that provide controlled drug release for weeks or months, improving treatment adherence and reducing dosing frequency.
- Polymer characteristics such as lactide-to-glycolide ratio, molecular weight, end-group chemistry, and glass transition temperature play a crucial role in determining degradation rate, drug release profile, and overall formulation performance.
- CROs employ multiple formulation platforms including microspheres, nanoparticles, and solid or in-situ forming implants, using technologies such as emulsion solvent evaporation, nanoprecipitation, spray drying, and hot-melt extrusion based on API properties.
- Advanced formulation optimization strategies help minimize the initial burst release through polymer selection, viscosity control, pore-sealing techniques, controlled solvent evaporation, and Design of Experiments (DoE) to achieve consistent product quality.
- Peptide stability challenges, including PLGA-induced acylation and acidic microenvironments, are addressed using ion-pairing, cationic salts, pH modifiers, and specialized polymer blends to preserve drug integrity during long-term release.
- Comprehensive analytical characterization using techniques such as FIB-SEM, X-ray microscopy, GPC, DSC, and Karl Fischer titration ensures Q3 microstructural equivalence, polymer consistency, and regulatory compliance for complex generic products.
- Successful commercialization depends on scalable manufacturing, robust Quality by Design (QbD), optimized in vitro release testing (USP Apparatus 4), moisture control, sterile processing, and regulatory expertise to deliver safe, effective, and FDA-compliant long-acting injectable therapies.

How Do Polymer Properties Influence PLGA Formulation Development Services?
The fundamental properties of PLGA polymers determine the degradation behavior and hydration kinetics of biodegradable matrices through factors such as molecular weight distribution, lactide-to-glycolide composition, and end-group chemistry. These characteristics collectively influence drug release kinetics, glass transition temperature, and the mechanical integrity of the final dosage form.
Developing a robust target product profile (TPP) within PLGA Formulation Development Services requires CROs to carefully evaluate and select suitable polymer grades based on their physicochemical characteristics. The composition and structural properties of the PLGA matrix serve as the primary determinants of controlled drug release. The most influential material attributes include:
Lactide-to-Glycolide (LA:GA) Molar Ratio:
Lactic acid (LA) contains a hydrophobic methyl side group, whereas glycolic acid (GA) lacks this substituent and exhibits greater hydrophilicity. As a result, PLGA with a 50:50 LA:GA ratio absorbs water more readily and undergoes faster hydrolytic degradation, typically breaking down within 3 to 4 weeks. Increasing the proportion of lactide, such as 75:25 or 85:15, slows water penetration and polymer erosion, making these compositions suitable for formulations designed to provide sustained drug release over several months.
Learn more about the technical impact of polymer selection: Understanding bulk erosion vs. surface erosion in PLGA.
Molecular Weight (Mw):
Polymers with higher molecular weights contain longer chains and a greater number of ester linkages that must undergo hydrolysis before the polymer fragments become sufficiently small to dissolve and erode. This characteristic delays bulk erosion and extends the duration of therapeutic drug release. In contrast, low molecular weight PLGA, typically ranging from 10 to 30 kDa, degrades more rapidly and provides faster drug release.
Compare degradation timelines: Analyze PLGA, PLA, and PCL degradation rates.
End-Group Chemistry:
Uncapped PLGA polymers terminate with hydrophilic carboxylic acid groups, commonly referred to as acid-terminated or acid-capped polymers. These terminal groups promote water uptake and accelerate the initial stages of polymer degradation. Ester-capped PLGA blocks these reactive terminal groups, reducing early hydration and slowing ester bond cleavage, thereby prolonging drug release.
Glass Transition Temperature (Tg):
The glass transition temperature (Tg) of PLGA generally ranges from 40°C to 60°C. This temperature marks the transition of the polymer from a rigid, glass-like state to a flexible, rubber-like state. During formulation development and storage, absorbed moisture or incorporated drug molecules may function as plasticizers, reducing the Tg and increasing polymer chain mobility, which subsequently accelerates drug diffusion.
For complex generic drug development programs, including abbreviated new drug applications (ANDAs), matching the polymer characteristics of the reference listed drug (RLD) is an essential regulatory requirement. A clear demonstration of this technical capability can be seen in the reverse engineering of commercially available long-acting depot formulations such as Lupron Depot and Vivitrol.
| Commercial Product / Reference Platform | Active Pharmaceutical Ingredient (API) | Polymer Composition (LA:GA Ratio) | Weight-Average Molecular Weight (Mw) | Number-Average Molecular Weight (Mn) | Polydispersity Index (PDI) | Glass Transition Temperature (Tg) | End-Group Termination / Microstructure Attributes |
|---|---|---|---|---|---|---|---|
| Lupron Depot (1-Month) | Leuprolide acetate (peptide) | 75:25 | ~13.0 kDa | ~8.7 kDa | ~1.5 | ~48.6°C | Acid-capped (acid number: ~12.9 mg KOH/g); contains gelatin |
| Vivitrol | Naltrexone (small molecule) | Approximately 50:50 | 83,118 ± 2,698 Da | 47,136 ± 1,145 Da | 1.89 ± 0.12 | 47.32 ± 0.19°C | Ester-capped; internal porosity: 51.59 ± 2.56%; residual moisture: 2.32 ± 0.13% |
Simplify your regulatory pathway: Discover our approach to PLGA polymer sameness for ANDA submissions.
What Processing Technologies Drive Microsphere, Nanoparticle, and Implant Programs?
The principal manufacturing technologies used in PLGA formulation development include emulsion-solvent evaporation (both single and double emulsion techniques), nanoprecipitation, spray drying, and hot-melt extrusion. The choice of manufacturing process depends on multiple factors, including the solubility characteristics of the active pharmaceutical ingredient, dosage requirements, thermal stability, and the intended duration of drug release.
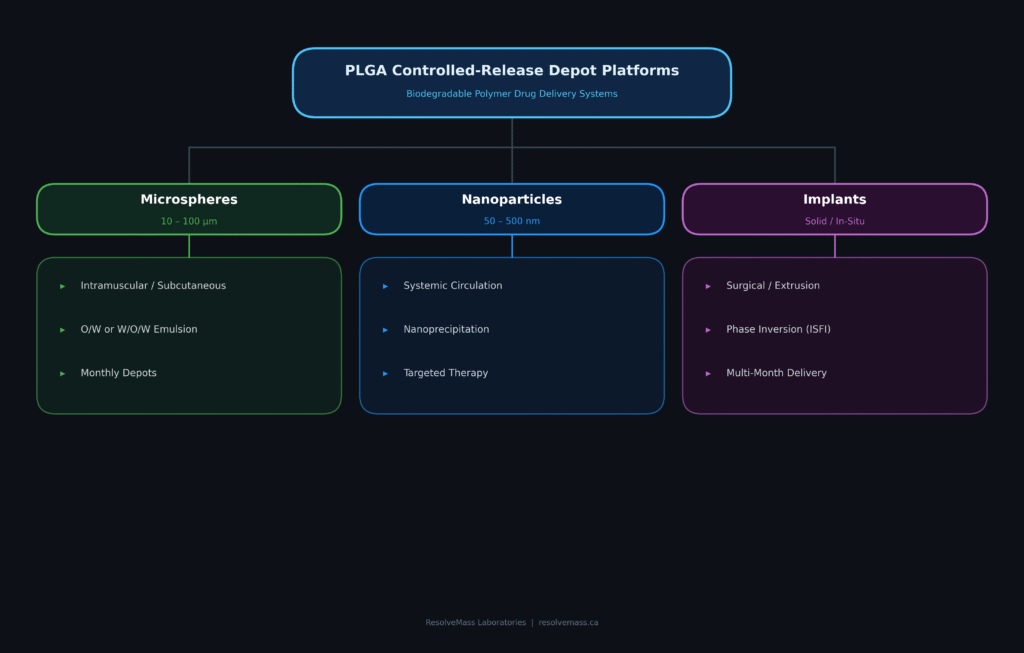
To accommodate the diverse characteristics of different API classes, CROs employ manufacturing technologies specifically optimized for each formulation platform.
Navigate manufacturing obstacles: Identify and overcome challenges in PLGA microsphere development.
Polymeric Microspheres
Polymeric microspheres are spherical delivery systems generally measuring between 10 and 100 µm in diameter and are intended for intramuscular or subcutaneous depot administration. The manufacturing strategy is primarily determined by the solubility profile of the incorporated API.
Optimize your formulation efficiency: Read about surfactants and emulsifiers in PLGA microsphere fabrication.
Single Emulsion (Oil-in-Water, O/W):
This technique is primarily used for hydrophobic drug molecules. Both the PLGA polymer and the drug are dissolved in an organic solvent, such as dichloromethane, before being emulsified into an aqueous phase containing a stabilizer such as polyvinyl alcohol (PVA). Subsequent evaporation of the organic solvent results in polymer solidification and microsphere formation.
Double Emulsion (Water-in-Oil-in-Water, W/O/W):
This method is specifically designed for hydrophilic APIs, including peptides and proteins. The drug is first dissolved within an internal aqueous phase and then emulsified into an organic phase containing dissolved PLGA to create the primary emulsion (W₁/O). This primary emulsion is subsequently dispersed into an external aqueous phase containing PVA to generate the secondary emulsion (W₁/O/W₂), followed by solvent extraction to produce solid microspheres.
Improve your encapsulation strategy: See methods for encapsulating hydrophilic vs. hydrophobic APIs in PLGA.
Polymeric Nanoparticles
Polymeric nanoparticles are submicron-sized delivery systems, generally ranging from 50 to 500 nm in diameter, making them well suited for intravenous administration, targeted drug delivery, and deep tissue applications. These carriers are commonly produced using nanoprecipitation, also referred to as solvent displacement. In this process, both the polymer and hydrophobic drug are dissolved in a water-miscible organic solvent, such as acetone, before being introduced gradually into an aqueous phase. Rapid solvent diffusion causes immediate precipitation of the PLGA polymer, resulting in the formation of ultra-fine nanoparticles.
Alternatively, advanced microfluidic manufacturing systems direct the polymer-containing organic phase and aqueous phase through precisely engineered micro-nozzles. This controlled mixing process enables highly reproducible nanoparticle synthesis, producing particles with exceptional size uniformity and a narrow polydispersity index (PDI).
Ensure high-quality particle production: Review the importance of controlling the PLGA PDI in pharmaceutical development.
Solid and In-Situ Forming Implants (ISFIs)
Solid PLGA implants are pre-manufactured cylindrical dosage forms produced using solvent-based processing or hot-melt extrusion (HME). These implants are typically administered surgically or through large-gauge injection needles.
In contrast, in-situ forming implants (ISFIs) rely on liquid phase inversion technology. In these systems, both the PLGA polymer and API are dissolved within a biocompatible, water-miscible solvent such as N-methyl-2-pyrrolidone (NMP) or dimethyl sulfoxide (DMSO). Following subcutaneous administration, the solvent gradually diffuses into the surrounding tissue fluids while water simultaneously penetrates the injected formulation. This solvent exchange triggers precipitation of the PLGA polymer, producing a solid depot directly at the injection site that enables sustained drug release over an extended period.
Explore specialized delivery sites: Learn about PLGA in CNS drug delivery and overcoming the blood-brain barrier.
How Does a CRO Mitigate the Initial Burst Phase in PLGA Formulation Development Services?
A contract research organization (CRO) minimizes the initial burst release by optimizing the viscosity of the organic phase, fine-tuning the polymer-to-drug ratio, and applying pore-sealing surface modification techniques such as PEGylation. These formulation strategies help reduce the premature diffusion of drug molecules positioned on or close to the outer surface of the PLGA polymer matrix.
The initial burst phase refers to the rapid release of drug molecules located near the particle surface or within interconnected, water-filled pores that are directly exposed to the external environment. If this release is not properly controlled, it can result in localized or systemic toxicity while simultaneously reducing the drug reservoir required to maintain sustained therapeutic concentrations over an extended period.
To quantitatively evaluate this phenomenon, formulation scientists commonly employ first-order kinetic models, including the Corrigan and Li equation, which specifically describes diffusion-controlled burst release:
Fburst = FBIN(1 − e−kbt)
- Fburst = Fraction of API released at time t
- FBIN = Total fraction of drug associated with the burst-release mechanism
- kb = First-order burst release rate constant
- t = Time
To effectively suppress the initial burst phase, CROs implement several formulation and process optimization strategies.
Pore-Sealing Through PEGylation
During solvent extraction, aqueous pore channels can develop within the polymer matrix, allowing hydrophilic drug molecules to escape rapidly. Incorporating a small percentage of PEG-capped PLGA, typically around 5% w/w, enables the PEG segment to migrate toward the droplet-water interface during emulsification. This creates a pore-sealing effect that reduces surface porosity and significantly limits the initial burst release.
Organic Phase Viscosity Optimization
Increasing the PLGA concentration within the organic phase, for example to approximately 10% w/v, increases solution viscosity. The higher viscosity prevents internal aqueous droplets from coalescing or migrating toward the microsphere surface during secondary emulsification, thereby improving drug encapsulation and reducing premature drug release.
Controlled Solvent Evaporation
Excessively rapid solvent removal promotes the formation of a rigid outer shell while simultaneously generating fractures and highly porous internal structures. Extending solvent evaporation over approximately four hours produces smoother, denser microspheres with substantially lower surface porosity, thereby minimizing burst release.
Peptide Salt Conversion
Hydrophilic peptide salts can be converted into more hydrophobic complexes, such as ion-paired pamoate salts, to reduce their aqueous solubility. Lower water solubility minimizes peptide migration into the external aqueous phase during emulsification, resulting in improved encapsulation efficiency and reduced initial drug loss.
Using structured Design of Experiments (DoE) methodologies, CROs systematically evaluate formulation and processing variables to establish an optimized manufacturing design space that consistently delivers the desired product characteristics.
| Process Variable Screened | Tested Range / Design Space | Optimized Set Point | Microstructural and Encapsulation Effect |
|---|---|---|---|
| Inner Aqueous Phase (W₁) Volume | 1%–10% v/v of the organic phase | 3% v/v | Minimizes internal water-droplet coalescence and increases encapsulation efficiency (EE) to greater than 72% |
| PLGA Concentration | 5%–20% w/v in DCM | 10% w/v | Increases continuous-phase viscosity, reducing peptide migration toward the oil-water interface |
| Organic Solvent System | Pure DCM vs. DCM/Ethyl Acetate (EtAc) blends | DCM:EtAc (3:1 v/v) | Optimizes solvent extraction kinetics while reducing surface defects and pore formation |
| Primary Homogenization Speed | 5,000–20,000 rpm | 10,000 rpm for 60 seconds | Produces a uniform primary emulsion droplet distribution without inducing peptide shear damage |
| PVA Stabilizer Concentration | 0.5%–5.0% w/v in W₂ | 1.0% w/v | Provides effective steric stabilization while preventing droplet coalescence and excessive surface adsorption |
| Hardening Bath Temperature | 4°C vs. Room Temperature (22°C) | 4°C | Reduces peptide partitioning into the external phase and lowers the initial 24-hour burst release to ≤8% |
| Peptide Salt Selection | Free acid vs. acetate vs. ion-paired pamoate | Pamoate salt conversion | Hydrophobic ion-pairing decreases peptide water solubility and increases baseline encapsulation efficiency by approximately 18% |
Master regulatory requirements: Achieve Q1/Q2 polymer equivalence assessment for your product.
What Are the Chemical Mechanisms and Mitigation Strategies for Peptide-PLGA Acylation?
Peptide acylation occurs when nucleophilic functional groups, including peptide amines, hydroxyl groups, or guanidine groups, react with the ester carbonyl groups present in degrading PLGA oligomers. To minimize this degradation pathway, CROs employ several stabilization strategies, including co-encapsulation of divalent cationic salts such as CaCl₂ and MnCl₂, hydrophobic ion-pairing techniques, and the incorporation of basic microenvironmental pH modifiers.
When peptides are encapsulated within degrading PLGA matrices, they are exposed to a highly dynamic and chemically complex microenvironment. As water penetrates the polymer, hydrolysis of ester bonds generates carboxylic acid-terminated oligomers. Because these acidic degradation products diffuse slowly through the hydrophobic polymer network, they accumulate within internal pores and lower the microenvironmental pH (μpH) to values approaching 2. This localized acidic environment accelerates autocatalytic polymer degradation while simultaneously producing reactive soluble oligomeric intermediates.
Peptide acylation proceeds through several distinct nucleophilic reaction pathways.
Primary Amine Acylation
Unprotonated amine groups located at the peptide N-terminus and on lysine side chains attack the carbonyl carbon atoms of degrading PLGA oligomers. This reaction produces stable covalent adducts that generate characteristic glycolyl mass shifts (+58.005 Da) or lactyl mass shifts (+72.021 Da). These modifications can be accurately identified using high-resolution liquid chromatography-mass spectrometry (LC-MS/MS).
Guanidine-Arginine Cyclization
For peptides that lack free N-terminal amines or lysine residues, the guanidine group of arginine becomes the primary reactive site. Following nucleophilic attack on PLGA carbonyl groups, spontaneous loss of ammonia (NH₃) occurs, producing stable cyclic derivatives including 2-oxazolin-4-one and 5-methyl-2-oxazolin-4-one. These products exhibit characteristic mass shifts of +41 Da and +55 Da, respectively.
Hydroxyl Esterification
Hydroxyl-containing amino acid residues, particularly serine, tyrosine, and terminal hydroxyl groups, can also react with PLGA carbonyl groups. This esterification process generates glycolic or lactic ester adducts that produce mass increases of +58 Da or +72 Da.
The overall kinetics of peptide acylation are governed by a competing equilibrium mechanism:
Peptide Amine Protonation (R-NH3+) ⇌ Reactive Nucleophile (R-NH2) + PLGA Sorption Precursor
(pH Dependent)
Under highly acidic conditions, where the microenvironmental pH approaches approximately 2, peptide amines become protonated, reducing their nucleophilic reactivity and theoretically slowing acylation. However, these positively charged protonated peptides exhibit strong electrostatic attraction toward negatively charged carboxylate end groups generated during PLGA degradation. This sorption effect concentrates peptides near highly reactive polymer surfaces, ultimately accelerating acylation despite the reduced nucleophilicity.
| Acylation Degradation Pathway | Targeted Nucleophilic Residues | Resulting Mass Modification | Directed CRO Stabilization Strategy |
|---|---|---|---|
| Primary Amine Acylation | N-terminal amines and lysine side chains | Glycolyl (+58.005 Da) or lactyl (+72.021 Da) adducts | Co-encapsulation of divalent cationic salts (CaCl₂, MnCl₂) |
| Arginine Guanidine Cyclization | Arginine residues, particularly in peptides lacking lysine (e.g., Goserelin) | Stable 2-oxazolin-4-one (+41 Da) or 5-methyl-2-oxazolin-4-one (+55 Da) derivatives | Blending with low molecular weight ester-capped PLGA polymers |
| Hydroxyl Esterification | Serine, tyrosine, and C-terminal hydroxyl groups | Glycolic or lactic ester adducts (+58 Da or +72 Da) | Reversible Hydrophobic Ion-Pairing (HIP) complexes |
| Acid-Catalyzed Hydrolysis | Labile peptide backbone bonds | Peptide chain fragmentation | Basic microenvironmental pH modifiers such as Ca(OH)₂ |
How Is Q3 Microstructural Equivalence Characterized in PLGA Formulations?
Q3 microstructural equivalence in PLGA formulations is established using advanced analytical techniques, including high-resolution focused ion beam scanning electron microscopy (FIB-SEM) and non-destructive X-ray microscopy (XRM) combined with quantitative AI-driven image segmentation. These technologies enable detailed mapping of the internal architecture of the formulation by identifying the spatial distribution of pores, polymer domains, and drug particles, allowing researchers to demonstrate structural equivalence with the reference product.
Recent Product-Specific Guidances (PSGs) issued by the FDA for complex long-acting formulations, including minocycline microspheres and dexamethasone inserts, have significantly increased regulatory expectations. Demonstrating bioequivalence is no longer limited to qualitative (Q1) and quantitative (Q2) sameness. It now also requires confirmation of microstructural (Q3) similarity. As a result, developers must compare the internal morphology, pore architecture, and phase distribution of the proposed generic formulation with those of the reference listed drug (RLD).
To achieve comprehensive Q3 characterization, CROs integrate high-resolution imaging techniques with advanced analytical methods to obtain detailed physical and chemical information.
FIB-SEM Imaging
Focused ion beam scanning electron microscopy (FIB-SEM) sequentially mills nanometer-scale layers from an individual microsphere or implant while scanning electron microscopy captures high-resolution cross-sectional images after each milling step. AI-assisted image segmentation then reconstructs these serial images into detailed three-dimensional models that accurately reveal the distribution of internal pores, polymer domains, and drug crystals throughout the formulation.
X-Ray Microscopy (XRM)
X-ray microscopy (XRM) is a non-destructive imaging technique that generates comprehensive microstructural datasets without damaging the sample. It enables visualization of both inter-particle and intra-particle density variations, allowing researchers to evaluate overall batch consistency and structural uniformity.
Gel Permeation Chromatography (GPC/SEC)
Gel permeation chromatography (GPC/SEC) is employed to determine the molecular weight distribution of both the raw PLGA polymer and the finished formulation. This analysis measures weight-average molecular weight (Mw), number-average molecular weight (Mn), and the polydispersity index (PDI), all of which are essential parameters for confirming polymer consistency.
Differential Scanning Calorimetry (DSC)
Differential scanning calorimetry (DSC) is used to determine the glass transition temperature (Tg) of the polymer while also identifying any polymorphic transformations that may occur within the encapsulated drug during formulation or storage.
Karl Fischer Titration (KFT)
Karl Fischer titration (KFT) accurately measures residual moisture content in PLGA formulations. Residual water must be maintained at extremely low levels, typically below 0.5%, to prevent premature hydrolytic degradation of the polymer during long-term storage and ensure product stability.
During reverse-engineering studies, the PLGA polymer must first be separated from excipients and the active pharmaceutical ingredient before comprehensive characterization can be performed. For example, when reverse-engineering the monthly leuprolide acetate depot formulation, free mannitol stabilizer is initially removed using ice-cold water. The remaining microspheres are then dissolved in tetrahydrofuran (THF), followed by molecular cut-off filtration to eliminate the peptide and gelatin stabilizers.
Following polymer isolation, titration procedures are performed to calculate the polymer’s acid number using the following equation:
Acid Number (AN) = (Vsample × NKOH × MWKOH) / WeightPLGA
The calculated acid number provides the absolute concentration of carboxylic acid end groups within the polymer, allowing investigators to determine whether the PLGA is acid-capped or ester-terminated.
What Are the Optimizing Parameters for Biorelevant USP Apparatus 4 In Vitro Release Testing (IVRT)?
Optimizing biorelevant USP Apparatus 4 in vitro release testing (IVRT) requires careful control of several critical parameters, including flow rate, flow-through cell geometry, dissolution media composition, temperature, and surfactant concentration. Proper optimization of these variables ensures that the IVRT method possesses sufficient discriminatory capability to detect formulation differences while providing meaningful predictions of in vivo drug release performance.
Developing a reliable IVRT method is essential throughout product development, routine quality control, and post-approval lifecycle management. The primary optimization parameters include the following.
Flow-Through Cell Hydrodynamics (USP Apparatus 4)
Standard USP Apparatus 4 systems typically utilize 22.6 mm flow-through cells packed with glass beads that distribute incoming media uniformly throughout the cell. This configuration minimizes particle compaction and prevents channel formation during testing. Standard operating conditions generally employ flow rates ranging from 3 to 17 mL/min under either open-loop or closed-loop configurations.
Real-Time Biorelevant Dissolution Media
Real-time IVRT studies are conducted under physiologically relevant conditions using 10 mM phosphate-buffered saline (PBS) maintained at pH 7.4 and 37°C. To preserve media integrity throughout multi-week release studies and prevent microbial contamination, antimicrobial additives such as 0.02% w/v sodium azide are commonly incorporated together with other stabilizing agents.
Accelerated Temperature and pH Conditions
To accelerate formulation screening and batch release testing, CROs frequently develop accelerated IVRT methods. Increasing the testing temperature to approximately 45°C–53°C enhances polymer chain mobility while accelerating ester bond hydrolysis. These conditions can reduce a conventional 30-day real-time release study to a 3-to-5-day accelerated assay without fundamentally changing the underlying release mechanism.
Maintaining Sink Conditions
Maintaining sink conditions remains one of the greatest challenges for APIs with poor aqueous solubility. To ensure continuous drug dissolution throughout the release study, dissolution media are optimized by adding surfactants such as 0.1%–1.0% sodium dodecyl sulfate (SDS) or Tween-80. These additives improve drug solubility without introducing membrane-controlled diffusion limitations that could alter release kinetics.
What CRO Capabilities Ensure Successful Process Scale-Up and Regulatory Approval?
Successful process scale-up and regulatory approval rely on CRO expertise in preserving geometric and kinematic similarity throughout scale-up, implementing comprehensive Quality by Design (QbD) principles, and validating robust sterile manufacturing processes. These capabilities enable a seamless transition from laboratory-scale formulation development to commercial current Good Manufacturing Practice (cGMP) production.
Transforming a laboratory-scale PLGA formulation into a commercially successful, FDA-approved long-acting injectable requires careful management of chemical, engineering, and regulatory considerations. Contract research organizations address several critical factors during this process.
Physicochemical Boundaries and Log P
Successful long-acting injectable (LAI) candidates generally require a monthly therapeutic dose of less than 100 mg to avoid excessive injection volumes and minimize injection-site discomfort. Drug molecules with a partition coefficient (log P) greater than 2.0 are particularly suitable because their hydrophobic nature facilitates efficient partitioning from the PLGA matrix into the surrounding tissue following administration.
Geometric and Kinematic Scale-Up
As manufacturing progresses from milligram-scale laboratory batches to multi-gram production, maintaining equivalent power-to-volume ratios and impeller tip speeds becomes essential. Changes in vessel geometry can alter solvent evaporation rates, ultimately affecting microsphere porosity and particle size distribution (PSD). These variations may significantly influence the magnitude of the initial burst release.
Purification and Moisture Control
Downstream purification commonly employs automated Tangential Flow Filtration (TFF) systems to remove residual organic solvents and excess polyvinyl alcohol (PVA) emulsifier. Following purification, optimized lyophilization cycles are validated to reduce residual moisture below 0.5%, thereby preventing premature hydrolytic degradation of the PLGA polymer during storage.
Container Closure and Stability Programs
CROs also perform comprehensive extractables and leachables (E&L) studies for elastomeric closures and prefilled syringe systems, validate container closure integrity (CCI), and conduct long-term and accelerated stability studies according to ICH guidelines. Typical stability conditions include 25°C/60% relative humidity for long-term storage and 40°C/75% relative humidity for accelerated stability assessments.
Maintain product integrity: Understand the factors influencing the shelf-life of PLGA, PLA, and PCL formulations.
Conclusion: Advancing Long-Acting Injectables via PLGA Formulation Development Services
Developing a comprehensive Chemistry, Manufacturing, and Controls (CMC) dossier demands expertise in polymer science, microencapsulation, and advanced microstructural analysis. Working with a specialized contract laboratory helps ensure that complex PLGA formulations meet regulatory bioequivalence requirements while maintaining consistent product quality and long-term stability.
Through the integration of polymer reverse engineering, Quality by Design (QbD) strategies, and detailed Q3 microstructural characterization, developers can reduce development risks and speed the path toward commercialization. PLGA Formulation Development Services deliver the technical capabilities, specialized processing expertise, and validation-ready analytical support required to successfully advance next-generation long-acting injectable therapies.
Stay ahead in biologics development: Explore our specialized characterization of long-acting biologics.
For technical questions or to discuss customized formulation development services, contact the team through the ResolveMass Contact Portal. To begin a reverse-engineering project or request cGMP polymer characterization services, connect with ResolveMass Laboratories through the ResolveMass Contact Page.
Frequently Asked Questions in PLGA Formulation Development Services
The terminal functional groups of a PLGA polymer have a significant impact on how quickly the material degrades after administration. Polymers with free carboxyl end groups absorb water more readily, which accelerates hydrolysis of the ester backbone. In contrast, ester-capped PLGA is less hydrophilic, reducing water penetration and extending the degradation and drug release period.
The W/O/W double emulsion technique is particularly suitable for water-soluble peptides because it isolates the drug within an internal aqueous compartment before encapsulation. This protective structure minimizes peptide loss during manufacturing and improves drug loading efficiency. As a result, it supports better encapsulation and more controlled release from PLGA microspheres.
Even small amounts of residual water can initiate slow hydrolysis of the PLGA polymer during storage, reducing molecular weight and potentially altering release performance. Excess moisture may also compromise product stability over time. To minimize these risks, optimized drying processes are employed, and residual water content is routinely monitored using Karl Fischer titration.
The glass transition temperature marks the point where PLGA changes from a rigid material to a more flexible polymer. Performing accelerated release studies at temperatures above this transition increases polymer mobility and water diffusion, allowing degradation and drug release to occur more rapidly. This approach significantly shortens testing time while preserving the underlying release mechanism.
Q3 microstructural equivalence is demonstrated by comparing the internal architecture of the test product with that of the reference formulation. Advanced imaging technologies such as FIB-SEM and X-ray microscopy are used to visualize pore networks, polymer distribution, and drug particle localization. These data help confirm that both products possess comparable internal structural characteristics.
As PLGA undergoes hydrolysis, reactive polymer fragments are generated that can chemically interact with susceptible peptide functional groups. Amino, hydroxyl, and guanidine residues are particularly vulnerable to these reactions, leading to the formation of modified peptide species. High-resolution analytical methods such as LC-MS/MS are commonly used to identify and quantify these degradation products.
Divalent salts such as calcium chloride and manganese chloride improve peptide stability by limiting interactions between peptide molecules and acidic PLGA degradation products. These ions compete for binding sites on the polymer, decreasing peptide adsorption within the matrix. Reducing this interaction lowers the likelihood of acylation and helps preserve peptide integrity during drug release.
Long-acting injectable formulations must remain suitable for administration through standard intramuscular or subcutaneous injections. Higher drug loads typically increase suspension viscosity and require larger injection volumes, making administration more difficult and less comfortable for patients. Keeping doses below approximately 100 mg helps maintain acceptable injectability and patient tolerability.
The ratio of lactic acid to glycolic acid determines the polymer’s hydrophilicity and rate of water absorption. Formulations containing a higher glycolic acid content generally absorb water more rapidly, resulting in faster polymer erosion and drug release. Increasing the lactic acid proportion slows degradation, making it suitable for formulations requiring extended release durations.
Reference:
- Campos-Morales, N., Cervantes-Pérez, L. G., Sánchez-Mendoza, A., Sánchez-Aguilar, M., Escobar-Chávez, J. J., Martínez-Acevedo, L., Galindo-Pérez, M. J., & Miranda-Calderon, J. E. (2026). PLGA-based in situ-forming implants, a quality by design perspective. Pharmaceutics, 18(3), 351. https://doi.org/10.3390/pharmaceutics18030351
- Hua, Y., Wang, Z., Wang, D., Lin, X., Liu, B., Zhang, H., Gao, J., & Zheng, A. (2021). Key factor study for generic long-acting PLGA microspheres based on a reverse engineering of Vivitrol®. Molecules, 26(5), 1247. https://doi.org/10.3390/molecules26051247
- Yang, J., Zeng, H., Luo, Y., Chen, Y., Wang, M., Wu, C., & Hu, P. (2024). Recent applications of PLGA in drug delivery systems. Polymers, 16(18), 2606. https://doi.org/10.3390/polym16182606
- Xin, H., Tan, S., Bandara, H. M. H. N., Fu, Y., Liu, S., & Smyth, H. D. C. (2014). Externally controlled triggered-release of drug from PLGA micro and nanoparticles. PLOS ONE, 9(12), e114271. https://doi.org/10.1371/journal.pone.0114271
- Hussain, O. R., & Lawan, R. L. W. (2024). PLGA implants for controlled drug delivery and regenerative medicine: Advances, challenges, and clinical potential. ResearchGate. https://www.researchgate.net/publication/391405437_PLGA_Implants_for_Controlled_Drug_Delivery_and_Regenerative_Medicine_Advances_Challenges_and_Clinical_Potential
- U.S. Food and Drug Administration. (2016). FY2016 regulatory science report: Long-acting injectable formulations. https://www.fda.gov/industry/generic-drug-user-fee-amendments/fy2016-regulatory-science-report-long-acting-injectable-formulations

