
Introduction
Effective Residual Solvent Control in PLGA Microsphere Manufacturing relies on optimizing post-formulation solvent removal processes and confirming regulatory compliance through validated headspace gas chromatography. This essential quality control strategy ensures that residual volatile organic solvents remain below established toxicological limits while maintaining the structural integrity and controlled drug release characteristics of the delivery system.
Need assistance optimizing your manufacturing process?Click here to learn about our PLGA long-acting injectable formulation services.
Poly(lactic-co-glycolic acid) (PLGA) microspheres are widely recognized as a fundamental platform for long-acting injectable depot formulations, enabling the sustained release of therapeutic agents over durations ranging from several weeks to multiple months. Throughout the manufacturing process, organic solvents play a vital role in dissolving the hydrophobic polymer matrix and facilitating the encapsulation of active pharmaceutical ingredients. Despite their importance during formulation, these volatile organic solvents provide no therapeutic value in the finished product and may present significant health hazards if retained above acceptable limits. Therefore, implementing comprehensive Residual Solvent Control in PLGA Microsphere Manufacturing is both a regulatory obligation and a clinical necessity. Residual solvents may adversely influence the physicochemical properties of PLGA microspheres by accelerating polymer degradation, triggering an excessive initial burst release of the drug, and reducing batch-to-batch consistency. As a result, manufacturers are required to design robust purification strategies that comply with the International Council for Harmonisation (ICH) Q3C guidelines while employing highly sensitive and validated analytical techniques to verify residual solvent levels and ensure product safety.
Ensure your product meets regulatory standards.Explore our expertise in PLGA polymer sameness for ANDA.
Share via:
Article Summary:
- Residual solvent control is essential in PLGA microsphere manufacturing because leftover organic solvents can affect patient safety, accelerate polymer degradation, alter drug release profiles, and compromise product consistency. Compliance with ICH Q3C and USP <467> guidelines is mandatory.
- Regulatory standards classify solvents by toxicity and establish Permitted Daily Exposure (PDE) limits. Manufacturers can demonstrate compliance using either Option 1 (fixed concentration limits) or Option 2 (maximum daily patient exposure), depending on the product formulation.
- Manufacturing conditions strongly influence solvent removal. Optimizing parameters such as solvent extraction rate, washing temperature, and diffusion kinetics helps prevent premature polymer shell formation that can trap residual solvents inside PLGA microspheres.
- Polymer vitrification is a major challenge during drying. As solvents are removed, the glass transition temperature (Tg) of PLGA increases, causing the polymer to become rigid and significantly slowing solvent diffusion. Understanding Tg behavior is critical for designing efficient drying processes.
- Advanced extraction technologies such as aqueous or alcoholic wet extraction and supercritical CO₂ extraction (SFEE) achieve much lower residual solvent levels than conventional vacuum drying while reducing processing time. However, extraction conditions must be carefully optimized to preserve microsphere structure and drug encapsulation efficiency.
- Residual solvents are typically measured using validated headspace gas chromatography (HS-GC) with FID or MS detection. Proper sample preparation, complete polymer dissolution, and method validation ensure accurate quantification and compliance with regulatory requirements.
- Successful residual solvent control requires an integrated approach that combines optimized manufacturing processes, a strong understanding of PLGA polymer behavior, and robust analytical testing to ensure product quality, regulatory compliance, and consistent performance of long-acting injectable formulations.

Regulatory Framework of Residual Solvent Control in PLGA Microsphere Manufacturing
The regulatory framework for Residual Solvent Control in PLGA Microsphere Manufacturing requires that residual volatile organic solvents comply with the stringent toxicological and exposure limits established by the ICH Q3C and USP <467> guidelines. These standards are implemented through well-defined solvent classifications and Permitted Daily Exposure (PDE) limits to minimize patient risk and ensure pharmaceutical safety.
Need help with characterization?Discover our services for the characterization of long-acting biologics.
The ICH Q3C guideline categorizes residual solvents according to their toxicological characteristics and defines acceptable exposure limits for each category. Organic solvents commonly employed during PLGA microsphere manufacturing must remain within the specified concentration limits or Permitted Daily Exposure (PDE) values to maintain regulatory compliance and protect patient health.
Classification and Concentration Limits of Common PLGA Process Solvents
Class 1 solvents are recognized as known or strongly suspected human carcinogens and environmental contaminants. Their use in pharmaceutical manufacturing should be avoided unless there is compelling justification based on a significant therapeutic advantage. Class 2 solvents consist of non-genotoxic animal carcinogens or compounds capable of producing reversible toxic effects, requiring their residual levels to be carefully controlled within prescribed limits. Class 3 solvents exhibit relatively low toxic potential and generally do not require stringent health-based exposure restrictions when present below established threshold values.
| Solvent | ICH Class | Permitted Daily Exposure (PDE, mg/day) | Concentration Limit (ppm) | Primary Manufacturing Role |
|---|---|---|---|---|
| Benzene | Class 1 | Avoided | 2 | Synthesis trace impurity |
| Carbon Tetrachloride | Class 1 | Avoided | 4 | Industrial chlorination trace |
| 1,2-Dichloroethane | Class 1 | Avoided | 5 | Synthesis trace impurity |
| Dichloromethane (DCM) | Class 2 | 6.0 | 600 | Polymer dissolution, emulsion formation |
| Acetonitrile | Class 2 | 4.1 | 410 | Extraction solvent, synthesis |
| Toluene | Class 2 | 8.9 | 890 | Synthesis of polymer intermediates |
| Methanol | Class 2 | 30.0 | 3000 | Washing, purification |
| 1,4-Dioxane | Class 2 | 3.8 | 380 | Emulsion phase modifier |
| Tetrahydrofuran (THF) | Class 2 | 7.2 | 720 | Reaction solvent, ring-opening polymerization |
| N-Methylpyrrolidone (NMP) | Class 2 | 5.3 | 530 | Polymer solvent, specialized depot vehicle |
| Ethylene Glycol | Class 2 | 6.2 | 620 | Synthesis byproduct, trace impurity |
| Ethyl Acetate | Class 3 | 50.0 | 5000 | Alternative green dispersion solvent |
| Acetone | Class 3 | 50.0 | 5000 | Solvent evaporation, precipitation |
| Ethanol | Class 3 | 50.0 | 5000 | Wet extraction washing, purification |
| Dimethyl Sulfoxide (DMSO) | Class 3 | 50.0 | 5000 | Active pharmaceutical ingredient solubilizer |
Regulatory Limits and PDE Evolution
The exposure limits established within regulatory guidelines are periodically reviewed and updated as new toxicological evidence becomes available. For instance, the allowable limit for Tetrahydrofuran (THF) was revised in the parent guideline to a Permitted Daily Exposure (PDE) of 7.2 mg/day, while N-Methylpyrrolidone (NMP) was assigned a reduced PDE of 5.3 mg/day based on updated toxicological evaluations.
An important historical example illustrating the evolving nature of regulatory guidance involves the Permitted Daily Exposure (PDE) for Ethylene Glycol. Before 2017, ICH Q3C Summary Table 2 identified Ethylene Glycol as a Class 2 solvent with a PDE of 6.2 mg/day (620 ppm). However, an inconsistency was observed because Appendix 5 listed a lower PDE of 3.1 mg/day. To address this discrepancy, the ICH Expert Working Group introduced a correction in the ICH Q3C(R7) revision, reducing the allowable limit to 3.1 mg/day. Subsequently, following a suspension request submitted in 2019, an extensive review of historical literature and the original 1997 toxicological assessment concluded that the previously established limit of 6.2 mg/day (620 ppm) was scientifically justified. Consequently, this value was reinstated in the currently applicable parent guideline, ICH Q3C(R6). This example highlights the importance of ensuring that analytical laboratories continuously monitor and implement the latest regulatory revisions to maintain ongoing compliance.
Mathematical Limit Calculations: Option 1 versus Option 2
The ICH Q3C guideline describes two approaches for determining the acceptable concentration limits of Class 2 residual solvents in finished pharmaceutical products.
Under Option 1, the concentration limit expressed in parts per million (ppm) is calculated using a standardized maximum daily product intake of 10 grams, according to the following equation:
Concentration (ppm) = (1000 × Permitted Daily Exposure (mg/day)) ÷ Dose (g/day) = (1000 × PDE) ÷ 10
When every active pharmaceutical ingredient and excipient complies with the Option 1 concentration limit, all formulation components may be combined in any proportion without exceeding the allowable patient exposure. However, formulations with lower daily doses or uneven solvent distribution among components may instead be evaluated using Option 2. Under this approach, the permissible solvent concentration is calculated based on the documented maximum daily dose of the final pharmaceutical product. Consequently, individual formulation components may exceed the Option 1 concentration limits, provided that the patient’s total cumulative daily exposure to the solvent remains below the established Permitted Daily Exposure (PDE).
The following example illustrates a representative pharmaceutical formulation containing one drug substance and two excipients, with a maximum daily dose of 5.0 g, evaluated for residual Acetonitrile (PDE = 4.1 mg/day; Option 1 limit = 410 ppm):
| Formulation Component | Amount in Formulation (g) | Acetonitrile Content (ppm) | Daily Solvent Exposure (mg) | Compliance Status |
|---|---|---|---|---|
| Drug Substance | 0.3 | 800 | 0.24 | Exceeds Option 1 limit |
| Excipient 1 | 0.9 | 400 | 0.36 | Meets Option 1 limit |
| Excipient 2 | 3.8 | 800 | 3.04 | Exceeds Option 1 limit |
| Total Drug Product | 5.0 | 728 (Weighted Mean) | 3.64 | Complies via Option 2 |
Although the Drug Substance, Excipient 2, and the final drug product all exceed the Option 1 concentration threshold of 410 ppm, the formulation remains fully compliant under Option 2 because the total daily patient exposure to Acetonitrile is 3.64 mg/day, which remains below the established Permitted Daily Exposure (PDE) of 4.1 mg/day.

Process Engineering and Optimization of Residual Solvent Control in PLGA Microsphere Manufacturing
Effective Residual Solvent Control in PLGA Microsphere Manufacturing depends on the careful optimization of critical process variables, including emulsion extraction rates, washing temperatures, and solvent diffusion kinetics. Proper control of these parameters minimizes the premature formation of an impermeable polymer shell that can entrap volatile organic solvents within the microsphere core.
Optimize your encapsulation techniques.Learn about encapsulating hydrophilic vs hydrophobic APIs in PLGA.
During emulsion-solvent evaporation or solvent extraction processes, such as W/O/W (water-in-oil-in-water) or O/W (oil-in-water) systems, solvent removal proceeds through partitioning of the organic solvent into the surrounding aqueous phase, followed by evaporation at the air-water interface. Halogenated solvents, particularly dichloromethane (DCM), possess very low water miscibility and relatively low boiling points, making them highly suitable for rapid emulsion formation. However, excessively rapid solvent extraction from the droplet surface causes the polymer concentration at the interface to rise sharply. This rapid desolvation promotes the immediate precipitation of localized polymer chains, resulting in the formation of a dense polymeric skin. Once established, this outer shell acts as a diffusion barrier, significantly restricting the migration of solvent molecules from the interior of the microsphere and trapping residual solvent within the core.
Understand degradation mechanisms.Explore bulk erosion vs surface erosion in PLGA.
To overcome this limitation, process engineers are increasingly investigating non-halogenated solvent systems, including ethyl formate, a Class 3 green solvent that offers favorable water miscibility and a low toxicological profile. Its improved extraction characteristics enable smoother solvent removal while maintaining excellent quality attributes in progesterone-loaded PLGA microspheres.
Vitrification and Glass Transition Dynamics in Residual Solvent Control in PLGA Microsphere Manufacturing
Vitrification is one of the most significant phenomena affecting Residual Solvent Control in PLGA Microsphere Manufacturing. During the drying process, the PLGA polymer transitions from a flexible rubbery state into a rigid glassy state, substantially reducing the free volume available within the polymer matrix. This transformation dramatically slows the diffusion of residual volatile organic solvents and presents a major obstacle to efficient solvent removal.
Need precise polymer analysis?Learn about PLGA PDI pharmaceutical characterization.
The glass transition temperature (Tg) defines the temperature range at which amorphous polymer chains change from a rigid glassy structure to a flexible rubbery structure. For dry PLGA copolymers, the baseline Tg generally falls between 30°C and 60°C, depending on several material characteristics, including the lactide-to-glycolide (LA:GA) ratio, molecular weight, and end-group chemistry.
| Polymer Grade | Lactide:Glycolide Ratio | End-Group Terminus | Baseline Tg (°C) |
|---|---|---|---|
| RG 502 S | 50:50 | Ester-terminated | 32.7 |
| RG 752 S | 75:25 | Ester-terminated | 35.8 |
| RG 755 S | 75:25 | Ester-terminated | 46.7 |
| RG 756 S | 75:25 | Ester-terminated | 49.8 |
| PLGA 503H | 50:50 | Acid-terminated | 46.0 |
During the desolvation process, residual solvent molecules function as highly effective plasticizers, causing a substantial reduction in the glass transition temperature (Tg) of the polymer-solvent mixture. This behavior can be described mathematically using the Gordon-Taylor equation:
Tg,mix = ( wpoly × Tg,poly + K × wsolv × Tg,solv ) ÷ ( wpoly + K × wsolv )
where wpoly and wsolv represent the weight fractions of the PLGA polymer and residual solvent, respectively, while Tg,poly and Tg,solv correspond to their individual glass transition temperatures. For example, the Tg of pure dichloromethane (DCM) is approximately 100 K (-173°C), whereas the Tg of water is approximately 138 K (-135°C). As these volatile components are progressively removed during drying, the glass transition temperature of the mixture (Tg,mix) continuously increases.
If the drying temperature decreases below the progressively increasing Tg,mix, the polymer undergoes vitrification and becomes a rigid glass. In this glassy state, polymer chain mobility is severely restricted, the available free volume decreases substantially, and the diffusion coefficient of the remaining volatile organic solvents declines by several orders of magnitude. Consequently, solvent removal becomes significantly slower, even under prolonged drying conditions.
Advanced Drying and Extraction Technologies to Accelerate Residual Solvent Control in PLGA Microsphere Manufacturing
Advanced drying and extraction technologies significantly improve Residual Solvent Control in PLGA Microsphere Manufacturing by employing wet alcoholic extraction techniques or supercritical fluid extraction (SC-CO₂) to preserve polymer free volume throughout the desolvation process. Compared with conventional vacuum drying, these approaches substantially reduce processing time while achieving much lower residual solvent concentrations.
Optimize your formulation process.Find out more about surfactants and emulsifiers in PLGA microsphere fabrication.
Traditional vacuum drying of solidified PLGA microspheres is relatively inefficient because vitrification restricts solvent diffusion through the polymer matrix. Comparative studies evaluating various desolvation methods have demonstrated that vacuum drying at 20°C reduces residual DCM only from approximately 5.0% to 4.34% after 18 hours. Increasing the vacuum drying temperature to 35°C improves solvent removal but still leaves a relatively high residual concentration of approximately 3.20%.
By comparison, aqueous wet extraction provides substantially greater solvent removal efficiency. At 20°C, residual DCM decreases to approximately 2.43%, while extraction at 35°C reduces the concentration further to approximately 0.03% (300 ppm). This enhanced performance occurs because water functions as a mild plasticizer, preserving sufficient free volume within the polymer network to facilitate continuous diffusion of DCM molecules out of the microspheres.
Additional improvements in desolvation efficiency can be achieved through redispersion of the microspheres in alcoholic media, such as aqueous solutions containing 20% to 50% methanol or ethanol. Alcohol penetrates the polymer matrix and acts as an effective plasticizer, increasing polymer chain mobility and promoting solvent diffusion. Using this strategy, residual DCM can be reduced from initial concentrations ranging between 4.0% and 7.0% to approximately 0.5%–2.3% within 1 hour, and further lowered to approximately 0.08%–0.18% (800–1800 ppm) after 6 hours of extraction.
Nevertheless, process engineers must carefully optimize both the alcohol concentration and extraction temperature. Excessive plasticization may soften the polymer matrix beyond acceptable limits, resulting in microsphere aggregation, structural deformation, and premature diffusion of the encapsulated drug.
Explore specialized applications.Learn about PLGA based ocular drug delivery systems.
The overall solvent removal pathway can be summarized as follows:
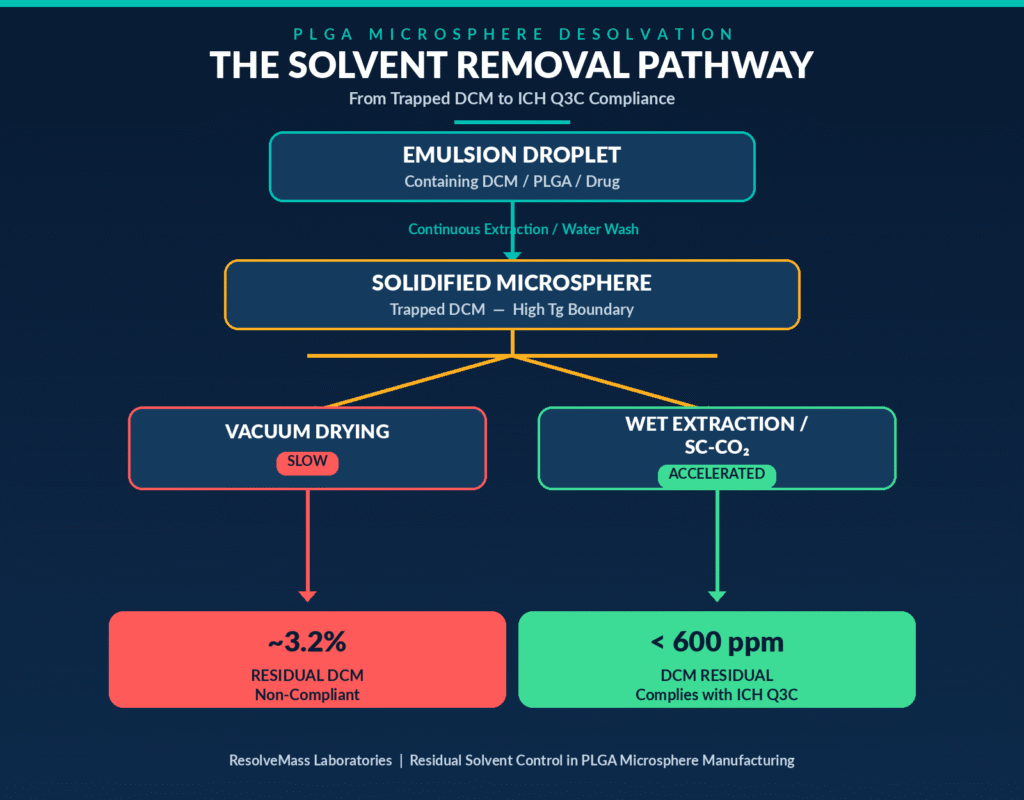
The thermodynamic and kinetic relationship between washing conditions and solvent removal is illustrated by comparative studies involving risperidone and naltrexone PLGA microspheres subjected to washing with 25% aqueous ethanol at different temperatures.
| Wash Temperature (°C) | Drug Class / Model | Residual Organic Solvent Content | Encapsulation Loading Efficiency (%) | Physical State of Microspheres |
|---|---|---|---|---|
| 20°C | Naltrexone Depot | 1.64% Benzyl Alcohol | 34.79 | Rigid, porous, no aggregation |
| 25°C | Naltrexone Depot | 0.74% Benzyl Alcohol | 34.72 | Stable, minor pore shrinkage |
| 30°C | Naltrexone Depot | 0.24% Benzyl Alcohol | 34.16 | Softened, minimal drug loss |
| 35°C | Naltrexone Depot | <0.05% Benzyl Alcohol | 28.00 | Softened, significant drug loss |
| 25°C | Risperidone Depot | <10 ppm DCM, 73 ppm EtOH | 37.52 | Smooth spherical structure |
| 30°C | Risperidone Depot | <10 ppm DCM, 176 ppm EtOH | 38.11 | High structural uniformity |
| 35°C | Risperidone Depot | <10 ppm DCM, 31 ppm EtOH | 37.21 | Minor structural deformation |
These findings clearly demonstrate the balance that must be achieved during process optimization. Increasing the wash temperature from 20°C to 35°C markedly enhances the removal of high-boiling residual solvents, such as benzyl alcohol, reducing their concentrations to nearly undetectable levels. However, the higher temperatures also soften the polymer matrix, facilitating outward drug diffusion and ultimately decreasing encapsulation efficiency. Successful process development therefore requires careful optimization of solvent extraction conditions to maximize residual solvent removal while preserving microsphere integrity and drug loading performance.
Supercritical Fluid Emulsion Extraction (SFEE)
An alternative strategy for enhancing solvent removal is Supercritical Fluid Emulsion Extraction (SFEE), which extracts organic solvents from O/W (oil-in-water) emulsions using supercritical carbon dioxide (SC-CO₂) within either a countercurrent packed column or a batch extraction vessel. This technique combines the excellent mass transfer characteristics of supercritical fluids with efficient solvent extraction, making it a promising approach for PLGA microsphere manufacturing.
In a representative investigation involving risperidone-loaded PLGA microspheres, batch extraction with SC-CO₂ was optimized using a Central Composite Design (CCD) to identify the operating conditions that maximized solvent removal efficiency. The optimized extraction parameters and resulting performance were as follows:
- Initial DCM Concentration: 4612 ± 198 µg/mL
- Optimal Static Extraction Time: 21.31 minutes
- Optimal Dynamic Extraction Time: 119.17 minutes
- Extraction Pressure: 226.44 bar
- Extraction Temperature: 44.77°C
- Final Post-Extraction DCM Concentration: <21.93 µg/mL, corresponding to an overall solvent removal efficiency of 99.43%
In addition to achieving exceptional residual solvent removal, SFEE produces microspheres with larger average particle sizes and increased internal porosity. These structural characteristics can improve the uniform distribution of the active pharmaceutical ingredient throughout the polymer matrix while maintaining desirable product quality attributes.
Analytical Testing Requirements for Residual Solvent Control in PLGA Microsphere Manufacturing
Quantitative assessment of Residual Solvent Control in PLGA Microsphere Manufacturing is typically performed using automated static headspace gas chromatography (HS-GC) coupled with either flame ionization detection (FID) or mass spectrometric (MS) detection. These analytical techniques provide the sensitivity, specificity, and quantitative accuracy required for regulatory release testing and routine quality control.
Static headspace sampling isolates volatile analytes from the non-volatile PLGA polymer matrix before chromatographic analysis. By preventing direct introduction of the polymer into the gas chromatograph, this approach protects both the injection system and the chromatographic column from contamination and fouling. During a standard analysis, the sealed headspace vial is heated to a controlled equilibration temperature, typically between 80°C and 105°C, while continuous agitation promotes thermodynamic equilibrium between the liquid sample phase and the gaseous headspace.
Need a comprehensive quality assessment?View our Q1-Q2 polymer equivalence assessment services.
Separation of residual solvents is commonly achieved using a bonded stationary phase composed of 6% cyanopropylphenyl and 94% dimethylpolysiloxane, corresponding to the USP G43 equivalent phase found in columns such as the Agilent DB-624 or Restek Rtx-1301. Although helium has traditionally been used as the carrier gas, modern analytical platforms employing nitrogen together with advanced autosamplers, such as the Thermo Scientific TriPlus™ 500 equipped with a direct column interface, can reduce sample equilibration times from approximately 60 minutes to 20 minutes. Under these optimized conditions, complete separation of Class 2A residual solvents can be achieved in less than 8 minutes, representing a substantial improvement over the conventional USP <467> analytical run time of approximately 60 minutes.
Sample Dissolution and Matrix Effect Mitigation
Accurate determination of volatile organic compounds requires effective mitigation of matrix effects through complete dissolution of hydrophobic PLGA microspheres in high-boiling, polar aprotic solvents such as dimethyl sulfoxide (DMSO) or dimethylformamide (DMF). Complete dissolution ensures uniform thermodynamic partitioning of volatile analytes into the gaseous headspace, thereby improving analytical accuracy and reproducibility.
When PLGA microspheres remain suspended without complete dissolution, residual solvents can become physically trapped within the polymer matrix, preventing them from reaching thermodynamic equilibrium with the headspace. This incomplete partitioning results in systematic underestimation of the true residual solvent concentration. Therefore, complete dissolution of the polymer matrix is essential to release entrapped volatile compounds before analysis.
Selection of an appropriate sample diluent depends on the solubility characteristics of the polymer, the physicochemical properties of the target analytes, and the possible presence of interfering volatile compounds.
Dimethyl Sulfoxide (DMSO):
DMSO is generally preferred for dissolving water-insoluble polymer systems because of its relatively low toxicity and high boiling point (189°C), which minimizes chromatographic background interference. However, because DMSO itself is non-volatile under standard headspace conditions, direct headspace analysis cannot be used to quantify residual DMSO. In such cases, direct liquid injection GC-FID using a suitable polar solvent, such as methanol, is the preferred analytical approach.
Dimethylformamide (DMF):
DMF is frequently employed for dissolving complex polymeric matrices and scaffold materials, particularly when quantifying high-boiling Class 2 solvents, including 1,4-dioxane. It provides excellent sample solubilization and minimizes matrix-related interferences while maintaining high analytical sensitivity. Because DMF is itself classified as a Class 2 residual solvent, appropriate laboratory safety procedures and contamination controls must be implemented during its use.
1,3-Dimethyl-2-imidazolidinone (DMI):
DMI is recommended as an alternative sample diluent when residual DMF or DMA is expected to be present in the sample, thereby preventing overlap between the diluent peak and the analytes of interest.
The composition of the sample matrix can influence the thermodynamic partition coefficient (K) of volatile analytes, leading to variations in detector response, chromatographic sensitivity, and baseline stability. Several advanced sample introduction techniques are available to minimize these matrix-related effects.
Multiple Headspace Extraction (MHE):
This method subjects a single sample vial to repeated cycles of equilibration and headspace injection. By mathematically modeling the exponential decline in analyte peak area over successive injections, the total analyte concentration can be accurately calculated, independent of matrix composition.
Purge-and-Trap Analysis:
In this technique, an inert carrier gas continuously bubbles through the dissolved sample, stripping volatile organic compounds from the liquid matrix. The released analytes are captured on an adsorbent trap, which is subsequently heated rapidly to desorb the compounds directly onto the GC column. Compared with conventional static headspace sampling, this approach can improve analytical sensitivity by as much as 1000-fold.
Because static headspace analysis involves pressurizing the sample vial before injection, daily fluctuations in ambient atmospheric pressure may introduce small variations in chromatographic retention times. To maintain quantitative consistency and analytical precision, each analytical sequence should include a certified standard reference mixture immediately following every sample analysis.
Method Validation and System Suitability Standards
Method validation and system suitability testing are fundamental requirements for demonstrating that analytical procedures consistently produce reliable and regulatory-compliant results. System suitability testing confirms adequate chromatographic performance through evaluation of critical peak resolution, injection repeatability, detector sensitivity, and overall instrument performance. These requirements must be fully validated according to ICH Q2 guidelines before the analytical method can be accepted for routine quality control or regulatory submission.
A validated analytical procedure must demonstrate acceptable specificity, linearity, accuracy, precision, robustness, and reproducibility. Prior to and throughout every analytical sequence, system suitability testing verifies that the chromatographic system continues to operate within predefined acceptance criteria.
Critical Pair Resolution
According to USP <467> and ICH recommendations, the chromatographic resolution (Rs) between the critical peak pair consisting of acetonitrile and dichloromethane should be at least 1.0, with a preferred value of 1.5 or greater for industrial applications. Chromatographic resolution is calculated using the following equation:
Rs = 2 × (tR2 − tR1) W1 + W2
where tR1 and tR2 represent the retention times of two adjacent chromatographic peaks, while W1 and W2 correspond to their respective baseline peak widths. High-efficiency chromatographic columns, including the DB-Select 624 UI, are capable of achieving resolution values approaching 3.1 for this critical solvent pair.
Signal-to-Noise Ratio (S/N)
System sensitivity verification requires that the signal-to-noise ratio (S/N) for 1,1,1-trichloroethane in the Class 1 standard solution be ≥5, while every peak contained within the Class 1 system suitability solution must demonstrate an S/N ratio of at least 3.
System Precision
Replicate injections (n = 5 or 6) of the reference standard solution must produce a Relative Standard Deviation (RSD) for peak area measurements of ≤10.0%, confirming satisfactory injection repeatability and detector precision.
Theoretical Plate Count
Chromatographic column efficiency, calculated from the analyte peaks of interest, should produce a theoretical plate count of at least 5000, ensuring adequate separation efficiency and sharp, symmetrical peak shapes.
All validation studies and supporting documentation should be compiled into a comprehensive validation package suitable for regulatory submissions, including IND, NDA, and ANDA applications.
Conclusion
Achieving effective Residual Solvent Control in PLGA Microsphere Manufacturing requires an integrated strategy that combines a comprehensive understanding of polymer phase-transition behavior, optimized solvent extraction processes, and rigorously validated chromatographic analysis.
A detailed understanding of polymer thermodynamics, particularly the influence of residual solvents on the glass transition temperature (Tg) of PLGA, enables the development of efficient drying protocols that minimize vitrification while maximizing solvent removal. At the same time, robust analytical methods validated according to USP <467> and ICH Q3C requirements ensure that residual volatile organic compounds are accurately identified and quantified, supporting both product quality and regulatory compliance.
ResolveMass Laboratories Inc. supports pharmaceutical innovators throughout every stage of this process by providing specialized analytical testing, dual GC-FID/GC-MS capabilities, and customized method validation services specifically designed for complex polymer matrices. Organizations seeking expert technical support or validated residual solvent analysis can contact the laboratory directly through the ResolveMass Contact Page.
Frequently Asked Questions
Residual solvents function as plasticizers within the PLGA polymer matrix by positioning themselves between polymer chains and increasing molecular mobility. This interaction lowers the glass transition temperature (Tg), making the polymer softer and more flexible than intended. If the Tg falls below physiological temperature, the microspheres may undergo premature structural changes, leading to accelerated polymer degradation and an undesirable burst release of the encapsulated drug.
The Gordon-Taylor equation is used to estimate how residual solvent content affects the glass transition temperature of a polymer-solvent system. By calculating the changing Tg during the drying process, manufacturers can determine suitable operating temperatures that promote efficient solvent removal without damaging the microsphere structure. This predictive approach helps prevent deformation, collapse, or instability while improving process control and product quality.
Vacuum drying frequently becomes less effective because rapid solvent evaporation at the surface causes the outer polymer layer to solidify much earlier than the interior. This phenomenon, known as vitrification, forms a dense outer shell that significantly slows the diffusion of trapped solvent molecules from the core. As a result, residual solvents may remain inside the microspheres even after extended drying periods, making it difficult to achieve regulatory limits using vacuum drying alone.
Wet extraction using aqueous mixtures containing ethanol or methanol enhances solvent removal by temporarily plasticizing the PLGA polymer matrix. This increases the free volume within the polymer and improves the mobility of trapped solvent molecules, allowing them to diffuse more efficiently into the extraction medium. As a result, residual dichloromethane (DCM) concentrations can be significantly reduced within a relatively short processing time compared with conventional drying techniques.
Supercritical Carbon Dioxide (SC-CO₂) combines gas-like diffusion properties with liquid-like solvent extraction capability, allowing it to penetrate deeply into the polymer matrix without causing structural damage. Under optimized operating conditions, it can remove organic solvents such as dichloromethane to extremely low concentrations while maintaining microsphere integrity. In addition, SC-CO₂ processing can improve particle porosity and promote a more uniform internal polymer structure.
Residual solvent analysis is commonly performed using capillary columns that comply with the USP G43 stationary phase specification. These columns contain 6% cyanopropylphenyl and 94% dimethylpolysiloxane and are typically available in lengths of 30 meters with internal diameters of 0.32 mm or 0.53 mm. Commercial examples include the Agilent J&W DB-624 and Restek Rtx-1301, both of which provide excellent separation of pharmaceutical residual solvents.
USP and ICH guidelines recommend achieving a chromatographic resolution (Rs) of at least 1.0, with a value of 1.5 or higher generally preferred for routine pharmaceutical analysis. Particular attention is given to the separation of the critical peak pair consisting of acetonitrile and dichloromethane. Adequate peak resolution prevents overlap during chromatography, allowing accurate peak integration and reliable quantification of individual residual solvents.
GC-MS is generally selected when samples contain unknown impurities, complex polymer matrices, or compounds that cannot be clearly distinguished using flame ionization detection alone. The mass spectrometer provides compound-specific fragmentation patterns that enable accurate identification through spectral library matching and selective ion monitoring (SIM). This higher level of selectivity makes GC-MS particularly valuable for complex pharmaceutical formulations and detailed impurity investigations.
Reference:
- Wang, Y., Bratlie, K. M., & Tang, L. (2020). Assessment of residual solvent and drug in PLGA microspheres by derivative thermogravimetry. Journal of Pharmaceutical Sciences, 109(10), 3085–3092. https://doi.org/10.1016/j.xphs.2020.07.005
- Kias, F., & Roland Bodmeier. (2024). Acceleration of final residual solvent extraction from poly(lactide-co-glycolide) microparticles. Pharmaceutical Research, 41(9), 1869–1879. https://doi.org/10.1007/s11095-024-03744-9
- Kattel, K., & Clogston, J. D. (2022). NCL method PCC-22: Residual organic solvent analysis in nanoformulations using headspace gas chromatography. In National Cancer Institute’s Nanotechnology Characterization Laboratory Assay Cascade Protocols. National Cancer Institute (US). https://www.ncbi.nlm.nih.gov/books/NBK604945/
- European Medicines Agency. (2024). ICH Q3C (R9): Residual solvents – Scientific guideline. https://www.ema.europa.eu/en/ich-q3c-r9-residual-solvents-scientific-guideline
- Osterberg, R. E. (2007, January). Impurities: Residual solvents [Conference presentation]. USP/PDA Residual Solvents Educational Conference, North Bethesda, MD, United States. https://www.uspnf.com/sites/default/files/usp_pdf/EN/USPNF/presentationosterberg.pdf
- U.S. Food and Drug Administration. (2017). Q3C: Tables and list—Guidance for industry. https://www.fda.gov/media/71737/download
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2019). ICH harmonised guideline Q3C(R6): Impurities—Guideline for residual solvents (2019 error correction). https://database.ich.org/sites/default/files/Q3C-R6_Guideline_ErrorCorrection_2019_0410_0.pdf

