
Introduction: Why Nitrosamines in Transdermal Patches and Topical Formulations Require Specialized Attention
Over the past several years, the pharmaceutical and regulatory sectors have invested significant effort into developing nitrosamine testing strategies primarily focused on solid oral dosage forms. However, nitrosamines in transdermal patches and topical formulations present a distinctly different and considerably more complex scientific and regulatory challenge. The matrices involved in these dosage forms, including pressure-sensitive adhesives, gel-forming polymers, permeation enhancers, and reservoir membranes, are chemically reactive, analytically difficult, and insufficiently represented in conventional analytical method libraries.
The global response to the 2018 valsartan recall initiated a major shift in nitrosamine oversight. Since that time, regulatory authorities have steadily expanded and strengthened expectations. The FDA’s Revision 2 guidance, issued in September 2024, significantly broadened the regulatory scope by formally distinguishing small-molecule nitrosamine impurities (SMNIs) from nitrosamine drug substance-related impurities (NDSRIs), while also requiring independent risk assessments for each category. For transdermal and topical drug products containing APIs such as rotigotine, rivastigmine, lidocaine, methylphenidate, nicotine, oxybutynin, and fentanyl, the difference between regulatory expectations and real-world analytical capability remains substantial. Identifying where these gaps exist, and implementing practical solutions to address them, has become a central concern for laboratories responsible for nitrosamine testing in these complex dosage forms.
To explore how these evolving regulatory requirements impact high-risk pharmaceutical categories, visit Resolve Mass Nitrosamine Testing for High-Risk Drug Classes.
Share via:
Article Summary:
- Nitrosamine evaluation in transdermal patches and topical dosage forms involves significantly more complex analytical hurdles than those typically encountered with conventional oral solid medications. These challenges are largely driven by difficult polymer and adhesive matrices, very low drug concentrations, and the current lack of detailed route-specific regulatory direction.
- Major regulatory authorities, including the FDA under its September 2024 Revision 2 guidance, EMA, Health Canada, and ICH M7(R2), expect manufacturers to follow a comprehensive three-step risk assessment strategy that addresses both small-molecule nitrosamine impurities (SMNIs) and nitrosamine drug substance-related impurities (NDSRIs).
- The Carcinogenic Potency Categorization Approach (CPCA) is used to establish acceptable intake (AI) limits for NDSRIs, with thresholds ranging from 18 ng/day up to 1,500 ng/day depending on structural potency classification. However, applying these exposure limits to transdermal delivery systems continues to be an area where regulatory expectations are still evolving.
- Among all analytical stages, sample preparation remains the most difficult and time-consuming component. Adhesive materials, penetration enhancers, solvents, and other formulation excipients can interfere with LC-MS/MS performance by affecting analyte extraction efficiency, signal recovery, and overall method sensitivity.
- Under FDA recommendations, confirmatory testing for drug products potentially containing NDSRIs, including transdermal systems, was expected to be completed by August 1, 2025. In comparison, Health Canada allows a longer CAPA implementation timeline extending through August 1, 2028 for acceptable intake limits issued before August 2025.
- Common nitrosamine mitigation strategies such as antioxidant incorporation, pH adjustment, and excipient qualification require extensive formulation-specific validation for transdermal products. Unlike conventional tablets or capsules, patch and topical systems may respond differently to these modifications due to their unique physicochemical and permeation characteristics.
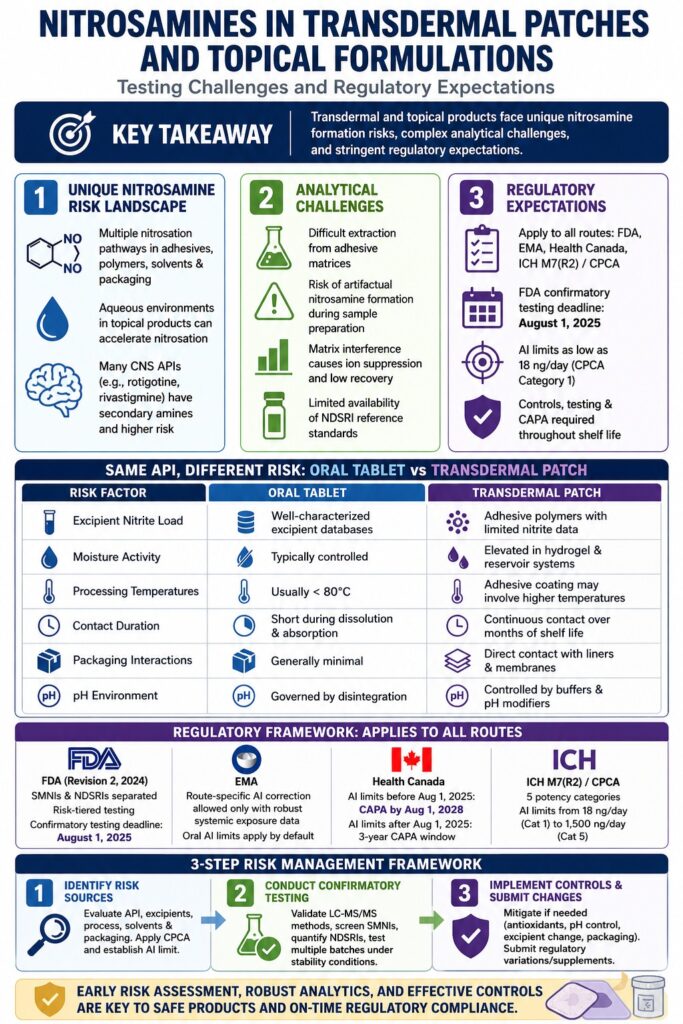
The Unique Nitrosamine Formation Risk Landscape in Transdermal and Topical Systems
Nitrosamine formation risks in transdermal and topical drug products differ fundamentally from those associated with oral dosage forms. Several formulation-specific and process-specific factors directly influence how risk assessments must be designed and interpreted.
Formulation-Specific Nitrosation Pathways
In transdermal patch systems, nitrosamine formation may originate from several distinct mechanisms:
- Secondary or tertiary amine-containing APIs reacting with trace nitrite impurities present in excipients, including cellulosic polymers, cross-linked polyvinylpyrrolidone, and certain adhesive systems that may retain residual nitrite from manufacturing processes.
- Residual nitrosating agents introduced through solvents used during adhesive coating operations. For example, dimethylformamide residues may contribute nitrosamine precursors under favorable conditions.
- Interactions involving packaging materials and release liners. Nitrocellulose-based release liners or printing inks may allow trace nitrite migration into the drug reservoir layer during long-term storage.
- Secondary amine functionality within APIs commonly used in central nervous system (CNS) transdermal therapies, such as rivastigmine, rotigotine, and methylphenidate. These APIs frequently fall into CPCA Category 2 or 3 NDSRI risk classifications.
For topical formulations such as creams, gels, ointments, and lotions, additional nitrosamine formation pathways may include:
- Preservative systems that place amine-containing excipients near nitrite-releasing compounds. Certain triethanolamine (TEA)-based emulsifiers are notable examples.
- Aqueous environments capable of accelerating nitrosation chemistry more readily than anhydrous transdermal systems.
- Antioxidant excipients that may unintentionally participate in nitrosamine-generating reactions under specific pH and temperature conditions.
For a deeper look into mitigating these risks using secondary amine scavengers, learn more at Resolve Mass Secondary Amine Scavenger Nitrosamine.
Why the Same API Can Carry Different Risk Profiles Across Dosage Forms
An important yet often overlooked consideration is that the same API may demonstrate dramatically different nitrosamine risks depending on the dosage form. An API considered low-risk in an oral tablet formulation may present substantially greater risk when incorporated into a transdermal patch system.
| Risk Factor | Oral Tablet | Transdermal Patch |
|---|---|---|
| Excipient nitrite load | Well-characterized excipient databases | Adhesive polymers with limited nitrite characterization |
| Moisture activity | Typically controlled through dry blending or granulation | Elevated in hydrogel and reservoir systems |
| Processing temperatures | Usually below 80°C | Adhesive coating processes may involve higher temperatures |
| Contact duration | Short during dissolution and absorption | Continuous contact over months of shelf life |
| Packaging interactions | Generally minimal | Direct contact with liners and membranes increases migration potential |
| pH environment | Governed by disintegration processes | Controlled by buffers and pH modifiers within the formulation |
These distinctions demonstrate why nitrosamine risk assessments developed for oral dosage forms cannot simply be transferred to transdermal or topical products without significant modification. Existing regulatory guidance still does not fully address these dosage form-specific complexities.
Regulatory Expectations for Nitrosamines in Transdermal Patches and Topical Formulations
Regulatory agencies currently apply a common three-step nitrosamine risk assessment framework across all pharmaceutical dosage forms. Nevertheless, route-specific guidance for transdermal and topical products remains limited.
FDA Guidance (Revision 2, September 2024): Implications for Non-Oral Dosage Forms
The FDA’s September 2024 Revision 2 guidance introduced the most substantial update to nitrosamine oversight since the initial 2020 framework. Several provisions directly affect transdermal and topical drug products:
- Formal separation of small-molecule nitrosamine impurities (SMNIs) from nitrosamine drug substance-related impurities (NDSRIs), each requiring distinct risk assessment workflows.
- A confirmatory testing deadline requiring manufacturers of approved products with potential NDSRI risk to complete testing and submit necessary regulatory changes by August 1, 2025.
- Explicit recommendations supporting reformulation approaches, including the use of antioxidants such as ascorbic acid and α-tocopherol, as well as pH modifiers intended to reduce NDSRI formation. For transdermal systems, these mitigation strategies must also demonstrate compatibility with adhesive properties and drug permeation performance.
- A risk-tiered testing strategy in which low-risk formulations may require only SMNI testing, while medium- and high-risk formulations require quantification of both SMNIs and NDSRIs.
One major unresolved issue is the absence of route-of-administration correction factors for transdermal and topical products. Until sufficient route-specific systemic exposure data become available, oral acceptable intake (AI) limits continue to apply to these dosage forms by default. This conservative approach may not accurately reflect true dermal absorption characteristics.
Review how international standards align on these guidelines at Resolve Mass Impact of ICH M7R2 Updates on Nitrosamine Risk Assessment.
EMA and the Route-of-Administration Challenge
Under EMA/409815/2020 and related Q&A guidance documents, the same nitrosamine risk assessment principles apply across all routes of administration. Adjustments to AI limits based on administration route are permitted only when the marketing authorization holder provides robust systemic exposure or bioavailability data specific to the transdermal route.
This issue has important implications for products such as:
- Rotigotine transdermal patches, where NDSRIs are included within EMA Appendix 1 classifications.
- Lidocaine topical products, which may generate NDSRIs due to secondary amine structures.
- Methylphenidate patches, which have already been discussed within EMA nitrosamine evaluation frameworks.
Without validated route-specific correction data, manufacturers must continue applying oral AI limits to transdermal systems. Since dermal bioavailability often ranges between 20% and 80% of oral exposure, these conservative limits can create technically difficult specifications for already complex matrix formulations.
To understand how these limits compare across global regulatory bodies, visit Resolve Mass Nitrosamine AI Limits Comparison.
Health Canada’s Timelines and CAPA Expectations
Health Canada follows a similar three-step nitrosamine evaluation process and maintains its own Appendix 1 list of established AI limits. For manufacturers of transdermal and topical products:
- AI limits published before August 1, 2025 permit CAPA implementation up to August 1, 2028.
- AI limits published after August 1, 2025 allow a three-year CAPA implementation period beginning from the publication date of the AI limit.
Health Canada also specifies that established AI limits apply through the end of shelf life. For transdermal patches with shelf lives extending 24 to 36 months, stability programs must therefore capture worst-case nitrosamine accumulation throughout long-term storage.
Learn how to execute long-term monitoring effectively by exploring Resolve Mass Nitrosamine Testing in Stability Studies.
ICH M7(R2) and the CPCA Framework Applied to Transdermal NDSRIs
The Carcinogenic Potency Categorization Approach (CPCA), now integrated into ICH M7(R2) discussions and adopted by major regulatory agencies, classifies nitrosamines into five potency categories:
| CPCA Category | Structural Basis | AI Limit |
|---|---|---|
| 1 (Highest potency) | Simple dialkyl nitrosamines such as NDMA | 18 ng/day |
| 2 | Limited α-hydrogens or activating structural features | 26.5 ng/day |
| 3 | Moderate α-hydrogen content | 100 ng/day |
| 4 | Presence of deactivating structural features | 400 ng/day |
| 5 (Lowest potency) | Strongly deactivating structural characteristics | 1,500 ng/day |
For transdermal APIs containing secondary amine functionality, Categories 2 through 4 are most frequently relevant. Achieving specifications of 26.5 ng/day or 100 ng/day within polymer-based patch systems is analytically much more challenging than meeting comparable limits in conventional oral tablets.
For insight into managing structural risks under this system, review the Resolve Mass Nitrosamine CPCA Approach for NDSRIs.
Analytical Testing Challenges Specific to Transdermal and Topical Matrices
Nitrosamine testing in transdermal patches and topical formulations presents analytical difficulties far beyond those encountered in solid oral dosage forms. In most cases, the matrix itself becomes the primary obstacle rather than the analytical instrumentation.
Sample Preparation: The Primary Technical Barrier
Sample preparation remains the single most critical factor determining success or failure in nitrosamine analysis for these dosage forms.
Adhesive Matrix Disruption
Pressure-sensitive acrylic and silicone adhesives commonly used in transdermal systems are poorly soluble in standard HPLC-compatible solvents. Complete extraction of trace-level NDSRIs from these adhesive networks often requires extensive solvent screening involving acetonitrile, methanol, tetrahydrofuran (THF), dichloromethane (DCM), or combinations thereof under varying temperatures and sonication conditions.
Poor and Variable Analyte Recovery
Adhesive polymers may physically entrap nitrosamine analytes, especially highly lipophilic NDSRIs. Recovery rates may vary significantly between manufacturing batches due to changes in polymer cross-linking density or nitrite contamination levels, which complicates method robustness and reproducibility.
Artifactual Nitrosamine Formation During Extraction
One of the most significant analytical concerns involves nitrosamine generation during sample preparation itself. If residual nitrites coexist with amine-containing APIs, extraction conditions involving acidic pH, elevated temperature, or prolonged contact time may artificially generate nitrosamines that were absent in the original product. This phenomenon can lead to false positive findings unless carefully controlled through optimized extraction conditions such as mild pH, low temperatures, and reduced extraction times.
Challenges in Topical Gels and Creams
Aqueous gels containing carbomer, hydroxypropyl cellulose, or xanthan gum introduce additional analytical complexity due to high viscosity. Creams and emulsions frequently generate biphasic extraction systems, while elevated water activity may support ongoing nitrosamine formation between sample preparation and instrument injection.
For an evaluation of optimal analytical techniques to overcome these extraction barriers, see Resolve Mass Direct Injection vs Headspace Techniques for Nitrosamines.
Instrumental Challenges: Sensitivity, Selectivity, and Matrix Effects
LC-MS/MS using triple quadrupole instrumentation remains the preferred platform for quantitative nitrosamine analysis at trace concentrations. However, transdermal and topical matrices create several instrument-specific complications.
Matrix-Induced Ion Suppression
Complex excipient systems containing adhesive polymers, penetration enhancers such as oleic acid, propylene glycol, and laurocapram, along with antioxidant additives, may significantly suppress electrospray ionization signals. This suppression can result in underestimation of actual nitrosamine concentrations. Matrix-matched calibration strategies and stable isotope-labeled internal standards (SIL-IS) are generally preferred corrective approaches, although SIL-IS materials remain unavailable for many NDSRIs.
API Mass Overlap with NDSRIs
NDSRIs typically differ from their parent APIs by approximately 29 Da due to incorporation of the nitroso group (-N=O). For large or structurally complex APIs, overlapping MRM transitions between the API and corresponding NDSRI may occur, requiring exceptionally strong chromatographic separation before MS detection.
Limit of Quantitation Constraints
Regulatory AI limits as low as 18 ng/day can translate into specification limits below 50 ng/g within patch matrices. Achieving validated limits of quantitation at these levels in highly complex transdermal systems requires advanced triple quadrupole instrumentation, highly optimized chromatography, and rigorous validation procedures.
Reference Standard Availability
For many transdermal-specific NDSRIs, including N-nitroso-rotigotine and N-nitroso-rivastigmine, commercially available reference standards may not exist. Without certified standards, analytical method validation cannot satisfy ICH Q2(R1) or Q14 requirements, confirmatory testing cannot proceed, and regulatory submission timelines may be jeopardized. As a result, custom synthesis of NDSRI reference materials should begin as early as possible, with development timelines commonly extending from three to six months.
Read about implementing high-sensitivity methods to meet these challenging limits at Resolve Mass Ultra-Low Limit of Quantitation (LOQ) in Nitrosamine Testing.
Risk Assessment Strategy for Transdermal and Topical Products: A Structured Framework
A robust and thoroughly documented risk assessment program is essential for regulatory compliance in nitrosamine management.
Step 1: Identify Nitrosamine Risk Sources
- Identify all secondary and tertiary amine functionalities within the API structure.
- Evaluate excipients, including adhesives, preservatives, penetration enhancers, and antioxidants, for nitrite, nitrate, and nitrosating potential.
- Assess manufacturing solvents and processing aids for dimethylamine or diethylamine contamination.
- Review packaging components, including liners, backing films, and pouches, for possible nitrite migration.
- Apply the CPCA framework to classify each potential NDSRI and establish the applicable AI limit.
Step 2: Conduct Confirmatory Analytical Testing
- Develop and validate LC-MS/MS methods specifically tailored to the transdermal or topical matrix.
- Perform SMNI screening for compounds such as NDMA, NDEA, NDIPA, NMBA, and NDBA where appropriate.
- Quantify identified NDSRIs associated with the API.
- Evaluate a minimum of three production-representative batches at both release and stability conditions to assess worst-case shelf-life accumulation.
Step 3: Implement Controls and Submit Regulatory Changes
- If nitrosamine levels exceed AI limits, implement mitigation measures including antioxidant incorporation, pH adjustment, excipient replacement, or process modifications.
- Submit regulatory variations or supplements in accordance with FDA, EMA, or Health Canada requirements.
- Confirm that all mitigation strategies preserve transdermal permeation performance, adhesive integrity, and overall product quality.
Discover specialized analytical solutions and method validation for complex frameworks at Resolve Mass Nitrosamine Analysis Services.
Formulation Mitigation: Key Considerations for Patch and Topical Systems
Mitigation approaches commonly used in solid oral dosage forms require careful modification when applied to transdermal and topical products.
Antioxidant Selection
Ascorbic acid and α-tocopherol (Vitamin E) are frequently used mitigation agents. However, their compatibility with adhesive systems and their impact on API permeation must be thoroughly evaluated. Due to its lipophilic properties, α-tocopherol may demonstrate greater compatibility with acrylic adhesive matrices compared to ascorbic acid.
pH Control
Many topical formulations already include buffering systems designed to maintain skin-compatible pH values between approximately 4.5 and 7.4. Lowering pH to suppress nitrosation reactions must be carefully balanced against skin tolerability and API stability considerations.
Excipient Substitution
Replacing excipients with elevated nitrite content or sourcing materials from suppliers with stricter nitrite controls can significantly reduce nitrosamine risk. Predictive modeling tools capable of estimating theoretical nitrosamine formation based on excipient nitrite levels should be incorporated early in formulation development for high-risk APIs.
Packaging Optimization
Improved packaging selection can substantially reduce oxidative and nitrosative stress throughout product shelf life. Replacing nitrocellulose-based liners and utilizing foil or aluminum pouch systems with low oxygen and moisture permeability are common risk reduction strategies.
Learn more about controlling risks introduced by your product primary packaging at Resolve Mass Packaging Leachables and Nitrosamine E&L Studies.
Conclusion: Bridging the Gap Between Regulatory Expectations and Analytical Reality
Nitrosamines in transdermal patches and topical formulations represent one of the most technically demanding areas in modern pharmaceutical quality and regulatory compliance. Regulatory expectations established through FDA Revision 2 (2024), EMA guidance, Health Canada initiatives, and ICH M7(R2)/CPCA frameworks clearly require all dosage forms, including complex transdermal and topical products, to undergo comprehensive nitrosamine assessment, testing, and control.
Despite this regulatory clarity, substantial operational challenges remain. The lack of route-specific AI corrections and the absence of validated analytical guidance specifically designed for adhesive and gel-based matrices continue to create significant technical barriers.
Laboratories and pharmaceutical manufacturers operating in this field require specialized expertise in NDSRI reference standard synthesis, transdermal matrix extraction optimization, LC-MS/MS method validation, and preparation of regulatory-compliant risk assessment documentation. Although the FDA’s August 2025 confirmatory testing deadline has passed, compliance activities remain ongoing, particularly with Health Canada’s CAPA implementation window extending through 2028.
At ResolveMass Laboratories Inc., our analytical scientists specialize in addressing these exact challenges, including custom NDSRI method development for complex transdermal matrices, ICH Q2(R1)-compliant validation programs, and comprehensive regulatory submission support for nitrosamine-related changes.
Contact the ResolveMass Laboratories Inc. team to discuss your transdermal or topical nitrosamine testing requirements.
Frequently Asked Questions (FAQs)
Yes. FDA nitrosamine guidance extends beyond oral dosage forms and includes all human pharmaceutical products, including transdermal patches, topical creams, gels, ointments, and lotions. Under the FDA’s Revision 2 guidance issued in September 2024, manufacturers are expected to conduct comprehensive nitrosamine risk assessments for both small-molecule nitrosamine impurities (SMNIs) and nitrosamine drug substance-related impurities (NDSRIs). The same three-step framework involving risk evaluation, confirmatory testing, and mitigation applies across all routes of administration. As a result, transdermal and topical products are subject to the same regulatory scrutiny as tablets and capsules.
At present, regulatory agencies such as the FDA and EMA do not provide standardized route-specific correction factors for transdermal or topical products. Manufacturers may propose adjusted acceptable intake limits only when supported by strong scientific evidence demonstrating compound-specific dermal absorption or systemic exposure differences. If no validated bioavailability data are available, regulators require companies to apply the same AI limits established for oral exposure. This conservative regulatory position often creates additional analytical and formulation challenges for transdermal systems.
Nitrosamine drug substance-related impurities (NDSRIs) are nitrosamine compounds formed directly from the API when amine-containing drug molecules react with nitrosating agents present within the formulation or manufacturing environment. APIs containing secondary or tertiary amine groups are generally considered higher risk for NDSRI formation. In transdermal and topical systems, APIs such as rotigotine, rivastigmine, methylphenidate, lidocaine, oxybutynin, and nicotine are frequently associated with elevated NDSRI concern because of their structurally reactive nitrogen functionalities. These compounds often require detailed risk assessments and targeted confirmatory testing.
Nitrosamine testing in transdermal patches is significantly more complex because adhesive polymers and topical excipients interfere with analytical performance. Acrylic adhesives, silicone matrices, penetration enhancers, and emulsifying agents can suppress ionization during LC-MS/MS analysis, making accurate quantification difficult at extremely low concentrations. In addition, extracting nitrosamines from adhesive matrices often produces inconsistent analyte recovery and may even generate artifactual nitrosamines during sample preparation. Achieving validated limits of quantitation below 50 ng/g in these formulations requires extensive method development, optimization, and matrix-specific validation strategies.
The Carcinogenic Potency Categorization Approach (CPCA) is a structure-based framework used by regulatory agencies to determine acceptable intake limits for nitrosamines based on their predicted carcinogenic potency. The system evaluates structural features such as α-hydrogen content near the nitroso group, as well as activating or deactivating substituents, to assign compounds into different potency categories. These categories correspond to AI limits ranging from 18 ng/day to 1,500 ng/day. For transdermal products, the same CPCA methodology is applied to NDSRIs, and oral AI limits remain the default unless reliable transdermal bioavailability data support an alternative limit.
The FDA recommended that manufacturers complete confirmatory testing for potential NDSRIs in approved pharmaceutical products, including transdermal patches and topical formulations, by August 1, 2025. This deadline also included submission of any required regulatory updates or product changes resulting from testing outcomes. Earlier deadlines were established for small-molecule nitrosamines, with completion expected by October 1, 2023. Although these deadlines have passed, ongoing compliance activities, additional testing, and corrective actions remain necessary for many products still under evaluation.
Many pharmaceutical excipients used in transdermal systems may contain trace nitrite impurities introduced during manufacturing or formed through degradation processes. Materials such as cellulosic polymers, polyvinylpyrrolidone, and certain adhesive grades are known potential sources. When these nitrites remain in prolonged contact with APIs containing secondary amine groups, nitrosation reactions may gradually occur during storage. Because transdermal patches maintain continuous API-excipient contact over extended shelf-life periods, even very low nitrite levels can become significant over time. Selecting low-nitrite excipient grades and qualified suppliers is therefore an important risk reduction strategy.
Yes. Topical creams, gels, and lotions often present a different nitrosamine risk profile than transdermal patches because of their higher water content and more chemically active environments. Aqueous systems can accelerate nitrosation reactions and may also contain preservatives, emulsifiers, or TEA-based compounds that introduce additional amine-nitrite interaction pathways. In contrast, many transdermal patches use relatively anhydrous adhesive matrices, which may reduce intrinsic nitrosation rates but create greater analytical extraction difficulties during laboratory testing. As a result, risk assessments must be tailored specifically to each dosage form.
A complete regulatory submission for nitrosamine management in transdermal or topical products typically requires several key components. These include a detailed written risk assessment covering APIs, excipients, manufacturing processes, and packaging materials, along with CPCA evaluations or compound-specific AI justifications for identified NDSRIs. Manufacturers must also provide validated analytical methods supported by ICH Q2(R1)-compliant validation data, confirmatory testing results from representative production batches, and stability studies demonstrating nitrosamine levels through the end of shelf life. Any implemented formulation changes, mitigation strategies, or process controls must also be fully documented with supporting validation evidence.
Reference:
- U.S. Food and Drug Administration. Control of Nitrosamine Impurities in Human Drugs — Guidance for Industry, Revision 2. September 2024. https://www.fda.gov/media/141720/download
- U.S. Food and Drug Administration. Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities (NDSRIs). CDER, August 2023. https://www.fda.gov/media/170794/download
- European Medicines Agency. Questions and Answers for Marketing Authorisation Holders/Applicants on the CHMP Opinion for the Article 5(3) Referral on Nitrosamine Impurities in Human Medicinal Products. EMA/409815/2020 (updated). https://www.ema.europa.eu
- Health Canada. Guidance on Nitrosamine Impurities in Medications. Updated 2024–2025. https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities.html
- ICH. M7(R2) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. Step 4 guideline, February 2023. https://database.ich.org/sites/default/files/ICH_M7(R2)_Guideline_Step4_2023_0216_0.pdf
- Nitrosamines Exchange (USP). Nitrosamines evaluation in transdermal patches — Risk Assessment Strategy/Tools & Technology thread. January 2024. https://nitrosamines.usp.org/t/nitrosamines-evaluation-in-transdermal-patches/9026
- ICH M7 Sub-Group Concept Paper. Principles of the ICH M7 Guideline to Calculations of Compound-Specific Acceptable Intake Limits. May 2024. https://database.ich.org/sites/default/files/ICH_M7SubGroup_Final_Concept_Paper_2024_0511.pdf

