
Bioanalytical CRO for Oncology Clinical Trials: Complex Matrices, PD Biomarkers, and Regulatory Expectations
Within the highly specialized field of oncology drug development, selecting an experienced Bioanalytical CRO for Oncology Clinical Trials plays a pivotal role in achieving successful regulatory approval. The analytical assessment of advanced oncology therapeutics, including antibody-drug conjugates (ADCs), cell and gene therapies, and bispecific antibodies, extends well beyond conventional plasma-based bioanalysis. These innovative therapies require the accurate evaluation of highly complex, non-liquid biological matrices alongside dynamic pharmacodynamic (PD) biomarkers. As pharmaceutical developers navigate significant biological variability together with continuously evolving regulatory requirements, collaborating with a specialized contract research organization becomes essential for maintaining data integrity and supporting clinical advancement. ResolveMass Laboratories Inc. meets these complex demands by integrating advanced analytical technologies with PhD-level scientific expertise to generate regulatory-quality data capable of meeting stringent international inspection standards.
To optimize drug development pipelines, many sponsors actively seek a trusted bioanalytical CRO partnership early in the design phase.
Share via:
Article Summary:
- Specialized bioanalytical CROs are essential for evaluating advanced oncology therapies using complex biological matrices and regulatory-compliant analytical methods.
- Accurate analysis of tissues, PBMCs, CSF, and whole blood requires advanced extraction techniques, LC-MS/MS, and flow cytometry to generate reliable PK and PD data.
- Pharmacodynamic biomarker validation relies on receptor occupancy assays, standardized PBMC processing, and sensitive multiplex platforms to assess immune response and treatment efficacy.
- FDA biomarker guidance and ICH M10 recommend a fit-for-purpose validation approach with robust statistical methods to ensure assay reliability and regulatory acceptance.
- Advanced therapies such as ADCs require multiple specialized assays to characterize drug behavior, stability, and safety throughout clinical development.
- Choosing an experienced, GxP-compliant bioanalytical CRO with strong regulatory expertise helps accelerate oncology drug development and supports successful global submissions.

Complex Matrices Evaluated by a Bioanalytical CRO for Oncology Clinical Trials
The analysis of non-liquid biological matrices, including solid tumor tissues, peripheral blood mononuclear cells (PBMCs), and cerebrospinal fluid (CSF), requires highly specialized sample preparation techniques designed to preserve analyte integrity while minimizing significant matrix-related interference. A dedicated Bioanalytical CRO for Oncology Clinical Trials must establish standardized workflows capable of consistently isolating, extracting, and accurately quantifying both therapeutic compounds and endogenous analytes from extremely limited clinical biopsy specimens.
Solid tumor tissues present substantial analytical challenges because of their considerable physical and biochemical heterogeneity. These tissues frequently contain varying proportions of necrotic regions, fibrotic stromal components, and highly vascularized margins, all of which can significantly influence localized drug distribution. Additionally, solid tissues are rich in intracellular lipids, structural proteins, and degradative enzymes that often contribute to pronounced matrix effects and ion suppression during chromatographic analysis. To overcome these obstacles, tissue-specific homogenization protocols combining mechanical disruption methods, such as bead-beating or pulverization, with carefully optimized chemical or enzymatic solubilization strategies must undergo thorough validation. In situations where authentic blank tissue samples are unavailable, which commonly occurs in rare oncology indications or brain biopsy studies, appropriately validated surrogate matrices must be developed to establish dependable calibration curves.
Sponsors targeting central nervous system malignancies depend heavily on dedicated tissue and CSF bioanalytical services to ensure accurate compartmental quantification.
Achieving lower limits of quantification (LLOQs) within the low picogram-per-milliliter range for these complex tissue matrices typically requires hybrid immunoaffinity liquid chromatography-tandem mass spectrometry (IA-LC-MS/MS). This advanced analytical approach utilizes antibody-coated beads or affinity columns to selectively capture the target biotherapeutic from tissue homogenates. Following capture, extensive washing procedures remove non-specifically bound endogenous proteins before instrumental analysis. Incorporating a dual-cycle immunoaffinity capture workflow with an acid dissociation step effectively disrupts drug-target complexes, significantly reducing matrix-derived interferences while substantially improving overall assay sensitivity.
Standardization of these complex non-liquid matrices is fundamental for accurately characterizing the pharmacokinetics (PK) of brain-targeted therapies and evaluating intratumoral drug distribution.
| Matrix Type | Primary Oncology Application | Common Extraction / Preparation | Primary Analytical Platform | Major Analytical Challenge | Mitigating Solution |
|---|---|---|---|---|---|
| Solid Tumors / Biopsies | Intratumoral drug distribution, target engagement, proteogenomics | Mechanical homogenization, enzymatic digestion | Hybrid IA-LC-MS/MS, MALDI-MS | Extensive tissue heterogeneity and significant matrix effects | Dual-cycle immunoaffinity enrichment with stable isotope-labeled internal standards |
| PBMCs | T-cell activation, immune checkpoint profiling, cellular PK | Density gradient centrifugation (Ficoll, SepMate, CPT) | High-Parameter Flow Cytometry | Rapid deterioration of cell viability and surface marker stability | Pre-optimized isolation tubes, strict cold-chain management, and accelerated sample processing |
| Cerebrospinal Fluid (CSF) | Glioblastoma studies, central nervous system (CNS) metastasis profiling | Protein precipitation, solid-phase extraction (SPE) | High-Sensitivity LC-MS/MS | Extremely low analyte concentrations and non-specific adsorption to collection containers | Anti-adsorptive tube treatment and sub-pg/mL sensitivity assays |
| Whole Blood | Receptor Occupancy (RO) assays, target-binding kinetics | Direct staining, fixative stabilization | Multiparameter Flow Cytometry | Limited post-collection stability window (24–48 hours) | Specialized fixatives and customized clinical sample collection kits |
At ResolveMass Laboratories Inc., senior scientific experts validate these multi-stage tissue extraction and preparation workflows using high-resolution mass spectrometry together with advanced chromatographic instrumentation. Through stringent quality control procedures applied throughout tissue homogenization and sample preparation, the laboratory minimizes analytical variability while consistently delivering reproducible, GxP-compliant datasets that support critical clinical development milestones.
Maintaining strict analytical control across these diverse sample types is fundamental to generating robust bioanalytical data throughout the trial lifecycle.
Validating PD Biomarkers with a Bioanalytical CRO for Oncology Clinical Trials
Successful validation of pharmacodynamic (PD) biomarkers in immuno-oncology studies requires sophisticated flow cytometry platforms, standardized peripheral blood mononuclear cell (PBMC) processing procedures, and multiplex analytical technologies specifically tailored to the intended context of use. High-parameter cellular analyses, combined with ligand-binding assays, allow clinical investigators to confirm target engagement, monitor immune system responses, and detect early indicators of therapeutic effectiveness.
Receptor Occupancy and Flow Cytometry Gating Strategies
Receptor Occupancy (RO) assays are among the most important tools used for pharmacodynamic monitoring in oncology clinical trials because they directly measure the interaction between monoclonal antibodies or bispecific biotherapeutics and their corresponding cell-surface targets. These assays are generally performed using three distinct analytical formats:
Free Receptor Assays: These assays quantify unoccupied cell-surface receptors using a fluorophore-conjugated detection antibody or drug analog that competes directly with the therapeutic agent for binding to the target receptor.
Occupied Receptor Assays: These assays directly measure receptors already bound by the therapeutic drug through the use of fluorescently labeled anti-drug antibodies (ADA) or non-competitive secondary detection reagents that specifically recognize the drug-receptor complex.
Total Receptor Assays: These assays determine the total density of cell-surface receptors by employing a non-competitive detection antibody that binds to a separate, non-overlapping epitope on the target receptor.
The percentage of receptor occupancy is mathematically determined using the relationship between free receptor signal intensity and total receptor signal intensity:
RO (%) = (1 − (Free Receptor Signal / Total Receptor Signal)) × 100
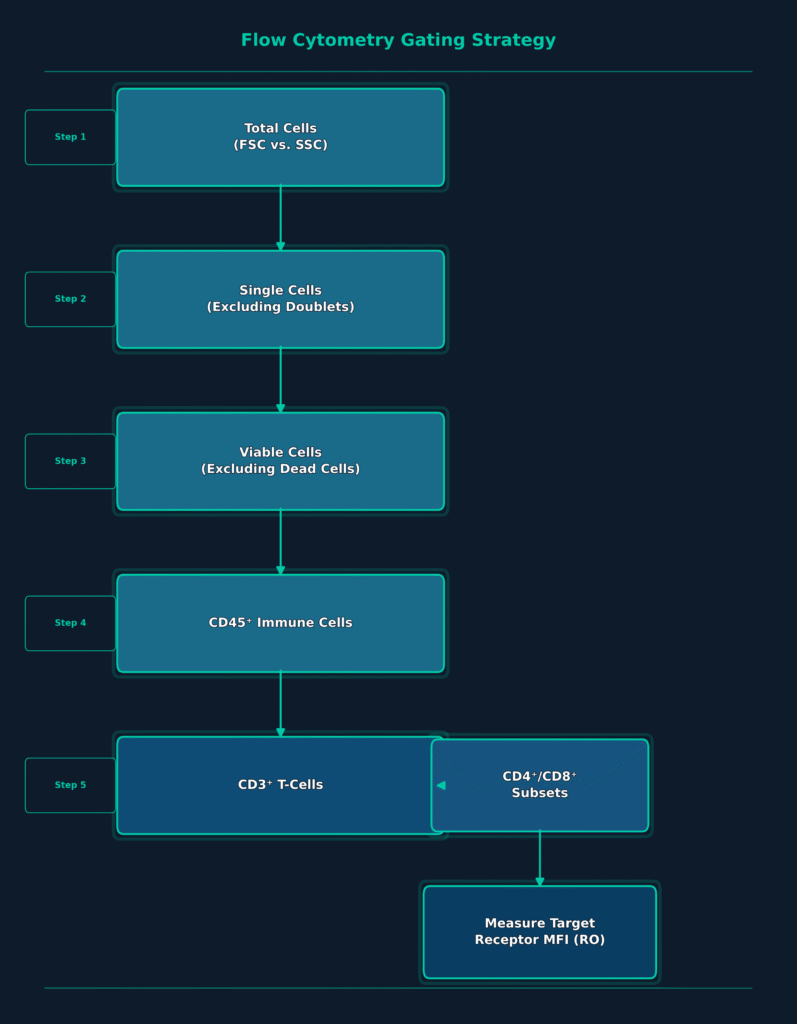
Reliable execution of receptor occupancy assays depends upon standardized multi-color flow cytometry gating strategies that accurately isolate the immune cell population of interest. A typical clinical gating workflow begins with forward scatter and side scatter (FSC vs. SSC) analysis to identify the overall cell population. Doublets are subsequently excluded to isolate individual cells suitable for downstream analysis.
A viability dye is then used to eliminate dead cells from the dataset, followed by application of the pan-leukocyte marker CD45 to distinguish immune cells from non-immune cellular populations. Sequential gating of lineage-specific markers, including CD3 for T-cells, CD4 and CD8 for helper and cytotoxic T-cell subsets, and CD20 for B-cells, further refines the target population. Finally, median fluorescence intensity (MFI) of the receptor or activation marker of interest, including PD-1, CD25, or HLA-DR, is measured within the selected cell population to determine receptor occupancy.
PBMC Processing and Multiplex Biomarker Platforms
Isolation of peripheral blood mononuclear cells (PBMCs) must be performed under GCLP-compliant conditions to preserve both cellular viability and biological function. The selection of the isolation system has a direct influence on sample quality and overall clinical trial performance.
CPT Tubes: These tubes contain a polyester gel together with density gradient medium, enabling direct PBMC separation during centrifugation at the clinical collection site. This approach simplifies sample handling while reducing logistical complexity and minimizing contamination risk.
SepMate Tubes: These specialized tubes incorporate a proprietary plastic insert that facilitates rapid and reproducible blood layering, enabling efficient PBMC recovery while requiring a manual pipetting step during processing.
Manual Ficoll Gradient Tubes: Although this method is cost-effective, successful PBMC isolation depends heavily on technician expertise, increasing the potential for variability in cell recovery across multiple clinical sites.
For fluid-based biomarkers, including cytokines, chemokines, and soluble immune checkpoint proteins, multiplex analytical platforms maximize the amount of biological information generated from limited clinical sample volumes. Meso Scale Discovery (MSD) electrochemiluminescence technology remains one of the preferred platforms for clinical biomarker validation because of its broad dynamic range and excellent resistance to matrix interference.
As biomarker candidates advance from exploratory research endpoints toward pivotal registrational biomarkers, many development programs transition to ultra-sensitive singleplex analytical platforms such as Single Molecule Array (Simoa) or automated Gyrolab systems. These highly sensitive technologies provide exceptional reproducibility while achieving detection limits in the sub-picogram-per-milliliter range, generating robust and reliable data to support critical clinical development decisions.
Meeting Regulatory Expectations as a Bioanalytical CRO for Oncology Clinical Trials
Complying with current regulatory expectations requires biomarker validation strategies to be aligned with the specific Context of Use (COU) while using the technical principles outlined in ICH M10 as the analytical foundation. On January 21, 2025, the U.S. Food and Drug Administration (FDA) published its final guidance document, “Bioanalytical Method Validation for Biomarkers,” formally distinguishing biomarker method validation from conventional pharmacokinetic (PK) bioanalytical assays. This regulatory milestone reflects the growing recognition that biomarker assays present unique scientific challenges requiring validation strategies that differ from those applied to traditional drug concentration measurements.
Sponsors must carefully align their assay protocols with the globally accepted ICH M10 bioanalytical method validation guidelines to satisfy international reviewers.
The Context of Use (COU) and Fit-for-Purpose (FFP) Paradigm
The 2025 FDA guidance emphasizes that biomarker assays should be validated according to a Fit-for-Purpose (FFP) framework driven by the study’s Context of Use (COU). The COU clearly defines the intended clinical application of a biomarker throughout the drug development process, whether it is used for patient stratification, pharmacodynamic safety monitoring, treatment response assessment, or as a primary efficacy endpoint. Unlike pharmacokinetic assays, which follow fixed and well-established acceptance criteria, biomarker assay performance characteristics, including precision, accuracy, and sensitivity, must be established according to the biological variability of the analyte and the magnitude of change required to support meaningful clinical decisions.
One of the most common errors observed during regulatory submissions is the inappropriate application of rigid, PK-focused ICH M10 validation criteria to complex endogenous biomarkers. Because biomarkers naturally occur within biological systems, they frequently lack an authentic reference material that perfectly matches the native analyte found in patient samples. Recombinant reference standards may exhibit substantial differences in glycosylation patterns, protein folding, or aggregation characteristics, making absolute quantification impractical or scientifically inappropriate.
As a result, enforcing conventional PK acceptance criteria may increase assay failure rates or produce misleading clinical datasets. Sponsors are therefore encouraged to engage regulatory authorities early in the development process and provide scientifically justified explanations for any departures from standard PK validation procedures within their method validation documentation. In addition, regulatory agencies recommend using the terms “validation” or “fit-for-purpose validation” instead of “qualification,” since the FDA reserves the term “qualification” exclusively for the biomarker itself as a drug development tool rather than the analytical assay used for its measurement.
Proactive identification of matrix variations helps teams anticipate and prevent unexpected bioanalytical method validation failure during late-stage assessments.
Implementing Statistical Cross-Validation Guidelines
Cross-validation performed under the ICH M10 framework should incorporate advanced statistical approaches, including Bland-Altman analysis and Deming regression, to evaluate systematic analytical bias instead of relying solely on traditional pass/fail acceptance criteria. Within the globally harmonized regulatory framework, cross-validation becomes mandatory whenever bioanalytical methods undergo significant modifications during long-term development programs or when clinical samples are analyzed across multiple laboratories.
When transitioning assays across geographic regions, a well-documented bioanalytical method transfer process ensures continued assay comparability.
| Statistical Tool | Primary Regulatory Application | Mathematical / Graphical Approach | Key Advantage over Classical Methods |
|---|---|---|---|
| Bland-Altman Plot | Visual assessment of analytical bias and agreement between laboratories or analytical platforms | Plots the difference between two analytical methods (Y₁ − Y₂) against their mean ((Y₁ + Y₂) / 2), using predefined equivalence limits | Detects systematic and concentration-dependent analytical bias throughout the entire measurement range instead of relying only on average correlation. |
| Deming Regression | Assessment of proportional and constant bias during cross-validation | Uses iterative regression modeling that accounts for measurement error in both the reference and comparison methods | Prevents the slope underestimation commonly associated with ordinary least squares (OLS) regression. |
| Lin’s Concordance | Evaluation of agreement relative to the 45-degree line of perfect concordance | Calculates the Concordance Correlation Coefficient (CCC), integrating both precision and accuracy | Measures both correlation and systematic deviation, preventing incorrect conclusions of equivalence when two methods are highly correlated but consistently offset. |
These statistical approaches are especially important in oncology clinical trials because of the highly dynamic interactions that occur between therapeutic agents and biological targets following drug administration. After dosing, patient samples typically contain varying proportions of free drug, drug-target complexes, and target receptors that exist in a continuously shifting equilibrium. Minor differences in laboratory temperature, incubation duration, or sample dilution procedures can significantly alter this equilibrium and introduce analytical bias.
Simple pass/fail acceptance criteria may fail to detect these subtle analytical discrepancies. Conversely, an integrated statistical strategy utilizing Bland-Altman analysis together with Deming regression can demonstrate that although baseline pre-treatment samples show excellent agreement across analytical laboratories, post-dose specimens may display systematic bias resulting from equilibrium shifts. This information enables researchers to implement scientifically justified mathematical correction factors before combining datasets for pharmacokinetic/pharmacodynamic (PK/PD) modeling.
Differentiating Parallelism from Dilutional Linearity in Oncology Assays
Parallelism evaluates the dilution-response behavior of endogenous analytes within their native biological matrix to confirm relative analytical accuracy, whereas dilutional linearity assesses serial dilutions of spiked reference standards prepared in a control matrix to identify hook effects and verify dilution integrity. Accurately distinguishing between these two validation parameters represents a fundamental aspect of bioanalytical method development.
Since biomarker assays are rarely calibrated using an authentic biological matrix identical to patient specimens, and instead commonly depend on surrogate matrices such as depleted serum or bovine serum albumin, demonstrating parallelism becomes one of the most important requirements for establishing quantitative assay reliability. Parallelism studies verify that the naturally occurring endogenous biomarker and the purified recombinant reference standard behave similarly during serial dilution, confirming that both are recognized equally by the assay antibodies.
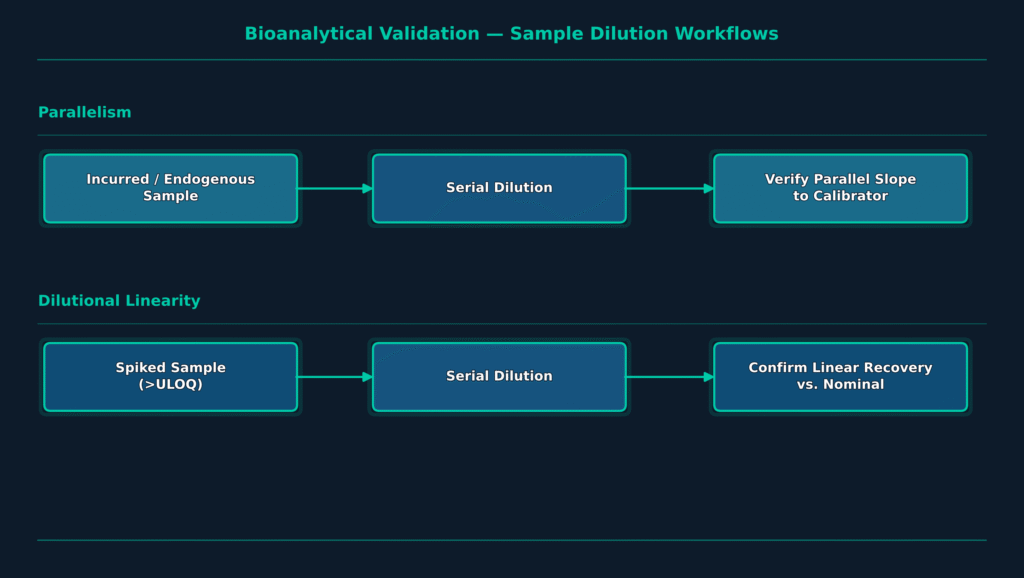
To assess parallelism under GCLP-compliant conditions, bioanalytical scientists typically evaluate three to ten individual samples collected from healthy individuals and patients with disease states that contain elevated endogenous concentrations of the biomarker. Each sample undergoes serial dilutions, most commonly using two-fold dilution steps, to produce a minimum of three measurable concentrations that remain within the validated quantitative range of the assay.
The resulting back-calculated, dilution-corrected concentrations are then compared across the dilution series. A lack of parallelism is generally concluded when the percent coefficient of variation (%CV) among all in-range measurements exceeds 30%. Parallelism studies also enable assay developers to establish the Minimum Required Dilution (MRD) needed to minimize endogenous matrix interference while defining the biological lower limit of quantification (LLOQ).
| Validation Parameter | Parallelism | Dilutional Linearity |
|---|---|---|
| Material Tested | Incurred clinical study samples containing elevated endogenous concentrations of the native biomarker. | Matrix samples fortified with purified recombinant reference standard. |
| Calibration Curve | Standard curve prepared using a surrogate analyte-free matrix. | Standard curve generated in the authentic biological matrix. |
| Primary Objective | Demonstrates equivalent dilution behavior between the endogenous biomarker and reference standard while establishing the MRD. | Evaluates assay recovery for samples exceeding the ULOQ and detects potential high-dose hook or prozone effects. |
| Acceptance Criteria | Dilution-corrected concentrations across serial dilutions must produce a %CV of ≤30%. | Measured concentrations must remain within ±15% of the nominal spiked concentration, consistent with ICH M10 recommendations. |
| Phase of Assessment | Initiated during early feasibility and method development, with additional verification following study completion when baseline concentrations are low. | Conducted during the pre-study method validation phase. |
In situations where acceptable parallelism is observed across only a limited section of the calibration curve, investigators may generate a Partial Parallelism Plot. This graphical analysis plots calculated concentration estimates against their respective dilution factors, allowing scientists to identify the precise concentration interval over which the assay maintains reliable analytical performance.
By defining regions of partial parallelism, assay developers can minimize the likelihood of generating inaccurate biomarker measurements while ensuring that clinical decisions are supported by statistically validated and scientifically reliable quantitative data.
Sponsors must also track technical variations across regional agencies, factoring in specific EMA vs FDA bioanalytical method validation differences to guarantee multi-regional acceptance.
Advanced Modality Testing by a Bioanalytical CRO for Oncology Clinical Trials
Developing comprehensive pharmacokinetic (PK) and pharmacodynamic (PD) profiles for emerging therapeutic modalities, including antibody-drug conjugates (ADCs), requires advanced multiplexed hybrid LC-MS/MS assays capable of simultaneously quantifying both conjugated and unconjugated molecular components. The intricate architecture of ADCs, consisting of a cytotoxic payload, a chemical linker, and a targeting monoclonal antibody, necessitates a sophisticated analytical approach to evaluate systemic stability, targeted drug delivery, and the potential for off-target toxicity throughout clinical development.
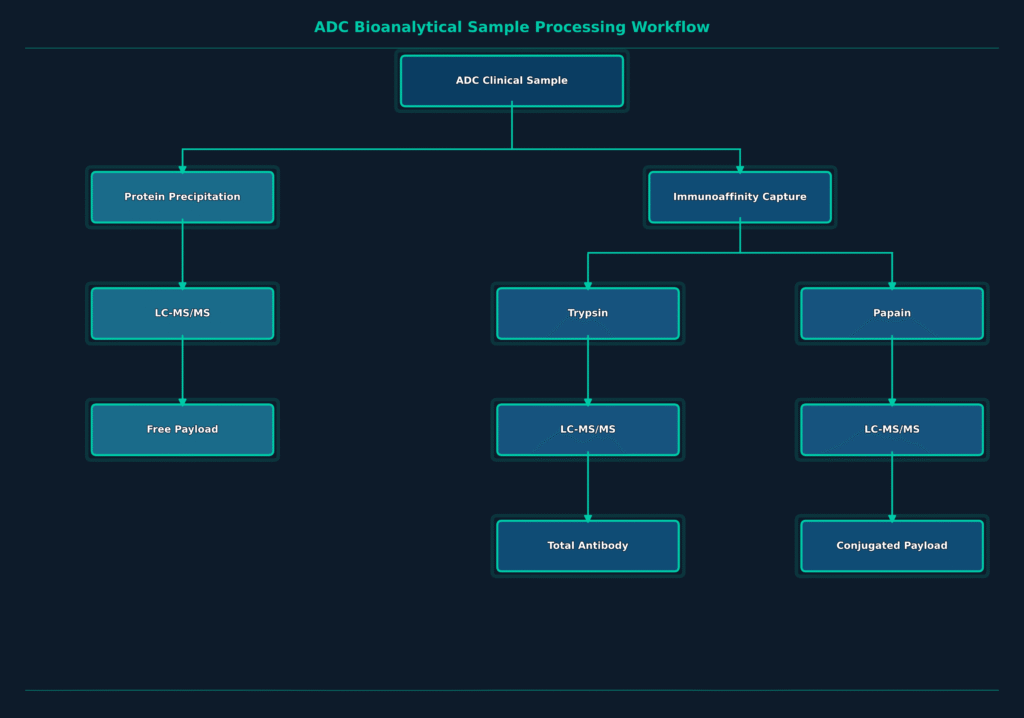
Accurate characterization of ADCs within biological matrices requires three separate quantitative assays, each providing essential information about different aspects of the molecule’s pharmacokinetic behavior.
Total Antibody Assay: This assay measures the combined concentration of both conjugated and deconjugated antibody species. The analytical workflow begins with immunoaffinity capture using anti-human IgG-coated beads, followed by trypsin digestion to generate a framework surrogate peptide suitable for subsequent quantification using LC-MS/MS.
Conjugated Payload (Active ADC) Assay: This assay determines the concentration of the cytotoxic payload that remains chemically attached to the antibody carrier. Following immunoaffinity capture of the ADC, enzymatic cleavage of the chemical linker, such as papain digestion for cleavable linker systems, releases the payload for highly sensitive LC-MS/MS quantification.
Free Payload Assay: This assay quantifies the concentration of cytotoxic small-molecule payload that has dissociated from the antibody during systemic circulation. The analytical procedure utilizes direct organic solvent precipitation to remove high-molecular-weight proteins, followed by high-sensitivity LC-MS/MS analysis capable of detecting free payload concentrations at low picogram-per-milliliter levels.
In addition to these quantitative assays, advanced bioanalytical laboratories routinely perform serum stability studies to determine the rate of payload deconjugation under physiological temperature and pH conditions over defined time intervals. These stability assessments, combined with high-resolution mass spectrometry measurements used to calculate the average drug-to-antibody ratio (DAR), provide critical information for predicting therapeutic index, evaluating molecular stability, and selecting the most promising clinical candidates.
As oncology drug development continues to expand toward innovative long-acting drug delivery systems, including poly(lactic-co-glycolic acid) (PLGA) and polylactic acid (PLA) microparticles, comprehensive characterization of polymer degradation and drug-release kinetics has become increasingly important. These biodegradable delivery platforms require specialized multi-detector gel permeation chromatography (GPC) techniques to accurately determine polymer molecular weight, polydispersity, and structural branching following in vivo administration.
Partnering with an integrated chemistry and bioanalytical CRO streamlines the process of evaluating both synthetic drug carriers and complex biological systems.
ResolveMass Laboratories Inc. remains at the forefront of these advanced analytical capabilities by combining custom peptide and polymer synthesis with state-of-the-art mass spectrometry technologies to support complex formulation development, analytical validation, and regulatory submission activities.
Selecting a Bioanalytical CRO for Oncology Clinical Trials
Achieving successful clinical trial outcomes depends on selecting a bioanalytical partner that combines advanced high-resolution mass spectrometry with comprehensive GxP-compliant data integrity systems. As international regulatory agencies continue to harmonize expectations under ICH M10 and the FDA’s January 2025 biomarker validation guidance, sponsors must ensure that their selected CRO demonstrates a well-established record of regulatory compliance, scientific excellence, and complete analytical data traceability.
Leading contract research laboratories maintain fully validated quality management systems supported by secure Laboratory Information Management Systems (LIMS) that comply with FDA 21 CFR Part 11 and EU GMP Annex 11 requirements. These systems protect raw chromatographic data, instrument records, audit trails, and clinical sample documentation from loss, unauthorized modification, or data integrity concerns. Equally important, the CRO should maintain complete lifecycle documentation for all critical assay reagents, including capture antibodies, calibration standards, and reference materials, to ensure consistent inter-batch reproducibility throughout long-term oncology development programs.
As oncology candidates advance to larger, multi-site programs, matching with an enterprise-ready bioanalytical CRO for Phase II and Phase III clinical studies is key to maintaining timeline momentum.
For biopharmaceutical organizations seeking the highest standards of scientific expertise and technical excellence, ResolveMass Laboratories Inc. serves as a trusted Bioanalytical CRO for Oncology Clinical Trials, supporting global product development and regulatory approval with comprehensive analytical solutions. Operating from its state-of-the-art facilities in Montreal, Canada, this USFDA-registered (Establishment Identifier No. 3042696771) and ISO 9001:2015-certified contract laboratory provides customized analytical method validation, impurity profiling, custom peptide synthesis, and polymer synthesis services. By partnering with ResolveMass, sponsors gain access to rigorous, reproducible, and regulatory-ready bioanalytical data that support the successful advancement of complex oncology therapeutics.
To discuss the specific bioanalytical requirements of your development program, establish a scientifically validated study strategy, or define timelines for GxP-compliant sample analysis, connect with a senior scientific expert at ResolveMass Laboratories Inc. today.
Frequently Asked Questions
A specialized Bioanalytical CRO for Oncology Clinical Trials is equipped with advanced analytical technologies, validated GxP quality systems, and scientific expertise to handle complex oncology samples and biomarkers. These organizations routinely analyze challenging biological matrices such as solid tumors, PBMCs, and cerebrospinal fluid using highly sensitive analytical methods. Their capabilities ensure reliable, reproducible data that meet stringent international regulatory standards while supporting every stage of oncology drug development.
Solid tumor biopsies are highly heterogeneous and contain varying amounts of lipids, proteins, fibrotic tissue, and degradative enzymes that can interfere with mass spectrometry measurements. These complex matrix components often cause ion suppression and reduce analytical sensitivity. To overcome these issues, bioanalytical laboratories implement validated tissue homogenization procedures together with hybrid immunoaffinity LC-MS/MS techniques that selectively isolate the target analyte before instrumental analysis.
Parallelism assesses whether endogenous biomarkers in patient samples respond consistently across serial dilutions, confirming that the native analyte behaves similarly to the calibration standard. Dilutional linearity, in contrast, evaluates the recovery of spiked reference standards after serial dilution to verify accurate quantification of samples above the assay’s upper limit of quantification (ULOQ). Together, these studies help ensure assay reliability and quantitative accuracy throughout clinical testing.
Peripheral blood mononuclear cells (PBMCs) are highly susceptible to changes in processing time, temperature, and sample handling conditions, making standardized validation essential. Proper isolation and handling preserve cell viability and functional integrity, both of which are critical for downstream immunological analyses. Consistent PBMC validation also improves the reliability of flow cytometry assays used to evaluate immune cell populations, checkpoint markers, and treatment-related cellular responses.
ICH M10 encourages the use of advanced statistical methods that evaluate analytical agreement and systematic bias instead of relying only on simple acceptance limits. Commonly used techniques include Bland-Altman plots to visualize differences between analytical methods, Deming regression to assess proportional and constant bias, and Lin’s Concordance Correlation Coefficient to measure overall agreement. These approaches provide a more comprehensive evaluation when comparing methods or laboratories.
A Bioanalytical CRO for Oncology Clinical Trials supports antibody-drug conjugate (ADC) development by generating detailed pharmacokinetic data for every major component of the molecule. Separate analytical methods quantify the total antibody, the conjugated cytotoxic payload, and the free payload released into circulation. Using immunoaffinity capture, enzymatic digestion, and high-sensitivity LC-MS/MS, these assays provide valuable information on ADC stability, drug release, and therapeutic performance.
Receptor Occupancy (RO) assays determine the proportion of target receptors occupied by a therapeutic agent on the surface of immune cells. This information helps establish the relationship between drug exposure and biological activity, providing valuable pharmacokinetic/pharmacodynamic (PK/PD) insights. RO data collected during early-phase clinical trials assist researchers in selecting appropriate dose levels, maximizing therapeutic benefit, and minimizing the risk of unnecessary toxicity.
Reference:
- Xue, Y.-J., Gao, H., Ji, Q. C., Lam, Z., Fang, X., Lin, Z., Hoffman, M., Schulz-Jander, D., & Weng, N. (2012). Bioanalysis of drug in tissue: Current status and challenges. Bioanalysis, 4(21), 2637–2653. https://doi.org/10.4155/bio.12.252
- National Center for Biotechnology Information. (n.d.). Bioanalytical challenges in drug measurement in tissue matrices. PubMed Central. https://pmc.ncbi.nlm.nih.gov/articles/PMC12291038/
- He, H., Yin, Y., & Chia, M. Y. (2016). Receptor occupancy assessment by flow cytometry as a pharmacodynamic biomarker in biopharmaceutical development. Clinical and Translational Science, 9(4), 209–217. https://doi.org/10.1111/cts.12399
- King, L., Allinson, J., Amaravadi, L., Kernstock, R., Garofolo, F., Gunsior, M., Jones, B., Mathews, J., Neely, R., Nelson, R., Pepin, M.-O., Shen, H., Stevenson, L., & Voelker, T. (2026). Best practices in the application of parallelism for biomarker assay validation. The AAPS Journal. https://doi.org/10.1208/s12248-025-01176-w
- National Center for Biotechnology Information. (n.d.). Record for PMID: 40021612. PubMed. https://pubmed.ncbi.nlm.nih.gov/40021612/
- Li, Z., Li, D. C., Yang, X., Liu, D., Wang, X., Yang, J., & Lu, Y. (2019). Validation and optimization of host immunological bio-signatures for a point-of-care test for tuberculosis disease. EBioMedicine, 40, 430–439. https://doi.org/10.1016/j.ebiom.2018.12.053

