
Introduction:
Establishing structural and functional similarity to an approved innovator biologic represents the cornerstone of regulatory approval for biosimilar products under abbreviated regulatory pathways worldwide. These regulatory pathways place significant emphasis on comprehensive analytical characterization and pharmacokinetic evaluations to confirm that no clinically meaningful differences exist between the biosimilar candidate and its reference biologic. This objective is achieved through carefully planned and scientifically rigorous Biosimilar Comparability Studies. This article examines the advanced scientific, regulatory, and statistical principles that guide the selection and sourcing of reference biological products, providing a comprehensive resource for biopharmaceutical developers seeking to strengthen and streamline global biosimilar development programs.
Need expert support for your comparability program? Explore our comprehensive Biosimilar Comparability Studies services.
Share via:
Article Summary:
- Reference product selection is critical for biosimilar approval, with global regulators increasingly accepting foreign comparators supported by robust analytical evidence.
- Using multiple reference lots from different batches and shelf-life stages helps capture natural variability and establish reliable comparability criteria.
- Effective sourcing requires overcoming challenges such as procurement costs, batch independence, and maintaining cold-chain integrity and full traceability.
- Advanced analytical techniques including mass spectrometry, peptide mapping, aggregate analysis, and receptor-binding assays are essential to confirm structural and functional similarity.
- Robust statistical methods, such as the Quality Range Maximum Likelihood (QRML) approach, improve the reliability of biosimilar similarity assessments.
- Successful biosimilar development depends on integrating sound reference product sourcing, advanced analytical characterization, statistical rigor, and regulatory compliance to support global approvals.
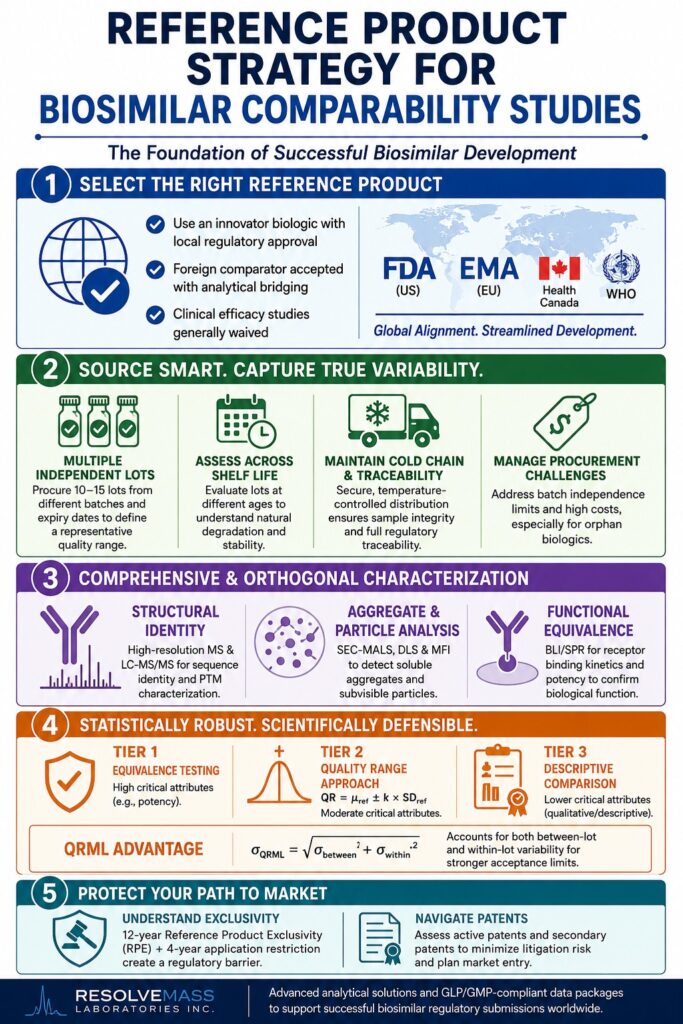
Global Regulatory Frameworks Governing Reference Product Selection
Across major regulatory jurisdictions, biosimilar guidelines require that the reference biological product used in comparability studies be an innovator biologic that has received local regulatory approval based on a complete, standalone clinical and quality dossier. This requirement ensures that the biosimilar candidate is evaluated directly against the established therapeutic standard that has already demonstrated safety, quality, and efficacy within the intended market.
Global regulatory expectations have become increasingly harmonized, particularly regarding the acceptance of foreign comparator products to facilitate more efficient international biosimilar development. A significant regulatory milestone occurred with the FDA’s March 2026 revision of its Biosimilar Q&A Guidance (Revision 4), which removed the default requirement for a mandatory three-way pharmacokinetic (PK) bridging study when developers utilize a non-U.S.-licensed comparator product. Under this updated regulatory framework, sponsors may justify the clinical relevance of data generated using a foreign comparator through comprehensive analytical evidence, including a Comprehensive Analytical Assessment (CAA), supported by strong scientific justification. This regulatory modernization has the potential to reduce pharmacokinetic development expenses by as much as 50%, allowing sponsors to save up to 20 million per development program by eliminating unnecessary clinical bridging studies.
Understand your regulatory requirements: Learn more about defining Critical Quality Attributes (CQAs) in biosimilars.
Likewise, Health Canada finalized its revised biosimilar guidance during late 2025 and 2026, confirming that comparative clinical efficacy and safety studies are “not typically required” when a biosimilar candidate can be extensively characterized and demonstrated to be highly similar through detailed structural, functional, and comparative pharmacokinetic evaluations.
The table below summarizes the current regulatory pathways and expectations regarding reference products across major international regulatory agencies.
| Regulatory Agency | Primary Assessment Pathway | Acceptability of Foreign Comparator | Clinical Efficacy Requirement |
|---|---|---|---|
| USFDA (United States) | 351(k) BPCI Act | Yes, with analytical bridging; default three-way PK bridging is no longer required as of March 2026 | Generally waived when robust analytical and PK/PD similarity has been demonstrated |
| EMA (European Union) | Centralised Procedure | Yes, provided the foreign comparator is authorized by an equivalent regulatory authority | Evaluated on a case-by-case basis using a tailored, evidence-based approach |
| Health Canada | New Drug Submission (NDS) | Yes, when the comparator is marketed in a jurisdiction with comparable regulatory standards | Not typically required; emphasis is placed on comparative PK, safety, and immunogenicity |
| WHO (World Health Organization) | Stepwise Biosimilarity Guidelines | Yes, with encouragement for mutual recognition and data-sharing among highly mature regulatory authorities | Supports tailoring clinical requirements according to the strength of analytical evidence |

Sourcing Strategy and Managing Originator Batch-to-Batch Variability
A robust sourcing strategy requires the procurement of multiple independent lots of the reference product obtained from different manufacturing batches and varying expiration dates to establish a statistically representative Quality Target Product Profile (QTPP). This approach enables developers to define realistic quality expectations and establish scientifically justified specifications that can withstand rigorous regulatory review.
Since biological medicines are produced using living host cell systems, they naturally exhibit inherent microheterogeneity that cannot be completely eliminated. Consequently, the reference biologic should not be viewed as a fixed chemical entity but rather as a dynamic target whose characteristics may gradually change over time because of post-approval manufacturing modifications introduced by the originator company.
Ensure high-quality data: Discover our specialized Biosimilar Characterization Services.
Sourcing Multiple Lots to Establish Acceptance Ranges
To develop a statistically sound analytical comparability program, biosimilar developers generally procure between 10 and 15 independent lots of the reference product. Evaluating only a small number of batches limits the ability to accurately define the complete range of the originator’s critical quality attributes (CQAs), potentially leading to quality specifications that are unnecessarily restrictive. When similarity limits are established using a limited and unrepresentative collection of reference lots, commercial biosimilar batches may experience increased rates of manufacturing non-compliance or unnecessary batch rejection during routine production.
In addition, the age of each reference lot at the time of analysis should be carefully considered. As biologic products approach their expiration dates, they naturally undergo degradation processes, including deamidation and aggregation. Assessing reference materials at different stages throughout their shelf life provides valuable insight into degradation pathways and the long-term stability profile of the target molecule, supporting more comprehensive analytical characterization.
Analyze degradation pathways: Read about forced degradation of biosimilars.
Overcoming Operational Pitfalls in Open-Market Procurement
Obtaining reference material directly from the originator manufacturer is rarely practical because innovator companies are generally unwilling to provide their products to potential competitors. As a result, biosimilar developers frequently depend on specialized open-market procurement channels to obtain commercial reference products. While this strategy is often necessary, it introduces several important operational and technical challenges.
Batch Independence Limitations
Large-scale biologic manufacturing frequently produces multiple finished drug product batches from a single large drug substance lot. Procuring several finished batches that originate from the same drug substance may artificially reduce the observed variability among reference samples, thereby weakening the statistical integrity of the analytical comparability assessment.
Orphan Biologic Procurement Costs
Obtaining reference products for orphan biologics presents a particularly difficult challenge. Because originator manufacturers maintain strict control over limited distribution networks and pricing is exceptionally high, procurement expenses can escalate rapidly. For example, eculizumab therapy may cost approximately 500,000 per patient each year, allowing procurement costs for reference materials alone to reach several million before analytical characterization even begins.
Cold-Chain and Traceability Integrity
Regulatory authorities require complete and verifiable traceability for every comparator lot included in biosimilar development studies. Sponsors must therefore establish secure, continuously temperature-monitored distribution systems that prevent thermal excursions capable of altering the physicochemical properties of reference materials. Failure to maintain cold-chain integrity can introduce analytical artifacts and potentially produce false-negative comparability results.
Advanced Mass Spectrometry and Orthogonal Characterization in Biosimilar Comparability Studies
Demonstrating a high degree of similarity in Biosimilar Comparability Studies requires a comprehensive panel of orthogonal analytical methods capable of evaluating structural, physicochemical, and biological characteristics. High-resolution mass spectrometry (HRMS), combined with liquid chromatography-tandem mass spectrometry (LC-MS/MS), remains the gold-standard analytical approach for confirming primary amino acid sequence identity and identifying post-translational modifications (PTMs).
Deep dive into structural analysis: Learn about Peptide Mapping in Biosimilars
and Post-Translational Modifications (PTMs) in Biosimilars.
At ResolveMass Laboratories Inc., analytical workflows integrate advanced mass spectrometry with state-of-the-art chromatographic technologies to achieve detailed site-specific characterization. Bottom-up peptide mapping verifies the precise amino acid sequence of both the variable and constant antibody domains while detecting trace-level point mutations, deamidation events, and oxidative modifications. Because conventional trypsin digestion can generate highly hydrophilic peptide fragments that exhibit poor retention on standard reversed-phase chromatographic columns, advanced characterization strategies incorporate complementary proteolytic enzymes such as Lys-C and Pepsin to achieve complete sequence coverage. In parallel, top-down and middle-down mass spectrometry approaches are employed to preserve and characterize labile PTM co-localization patterns that may otherwise be lost during enzymatic digestion.
Advanced chromatographic technologies, including Waters MaxPeak High Performance Surfaces (HPS), further enhance analytical performance throughout these workflows. By minimizing non-specific interactions between peptides and metal surfaces, HPS technology significantly reduces peak tailing while improving the recovery of acidic and phosphorylated peptides by as much as 34-fold. These improvements contribute to highly reproducible peptide mapping and greater confidence in structural characterization results.
Explore advanced mass spec techniques: Read about Native Mass Spectrometry for Biosimilars.
Orthogonal Methods for Aggregate and Particle Characterization
No single analytical technique is capable of characterizing the complete size spectrum of therapeutic protein aggregates. Consequently, comprehensive aggregate assessment requires an orthogonal analytical platform that combines Size-Exclusion Chromatography coupled with Multi-Angle Light Scattering (SEC-MALS), Dynamic Light Scattering (DLS), and Micro-Flow Imaging (MFI). Together, these complementary methods enable developers to evaluate soluble oligomers, hydrodynamic particle sizes, and subvisible particles with high sensitivity, even at trace concentrations.
Manage product stability: Understand our approach to Aggregation Analysis in Biosimilars.
ResolveMass Laboratories Inc. employs qualified, GLP-aligned analytical workflows to comprehensively assess aggregate size variants and characterize the morphology of subvisible particles. Size-Exclusion Chromatography coupled with Multi-Angle Light Scattering (SEC-MALS) provides absolute molecular weight measurements without relying on column calibration, making it a highly reliable technique for aggregate characterization. Dynamic Light Scattering (DLS) serves as a rapid, low-sample-volume screening method that identifies early-stage aggregation behavior in solution. Complementing these approaches, Micro-Flow Imaging (MFI) passes liquid samples through a high-resolution imaging system that captures digital images of individual particles, enabling both quantitative particle counting and detailed morphological assessment of subvisible particles ranging from 1 to 100 microns. This morphological evaluation effectively differentiates proteinaceous aggregates from air bubbles, silicone oil droplets, and glass delamination particles, thereby minimizing the likelihood of regulatory concerns arising from aggregate misidentification.
Assess molecule heterogeneity: Learn about Glycosylation Analysis of Biosimilars
and Charge Variant Analysis in Biosimilars.
Functional Characterization and Receptor Binding in Biosimilar Comparability Studies
Potency assays and receptor-binding kinetic analyses play a critical role in demonstrating that the structural similarity of a biosimilar translates into equivalent biological function and comparable in vivo pharmacokinetic performance. For monoclonal antibodies, a comprehensive characterization assay (CAA) panel generally includes assessments of antigen-binding affinity, C1q interaction, and a complete panel of activating and inhibitory Fcγ receptor interactions.
Bio-Layer Interferometry (BLI) and Surface Plasmon Resonance (SPR) are widely utilized to measure receptor-binding kinetics in real time. During FcRn kinetic studies, which are designed to predict an antibody’s serum half-life, the experimental protocol must accurately replicate the pH-dependent recycling mechanism. This requires monitoring antibody binding to the FcRn receptor at the acidic endosomal pH of 6.2, followed by rapid dissociation at the physiological pH of 7.4. To maintain assay sensitivity and minimize kinetic artifacts associated with fast association and dissociation receptors such as CD32a/FcγRIIa, developers should optimize biosensor loading densities. For example, utilizing a His XT biosensor loaded to less than 10% of its maximum capacity, corresponding to a signal shift below 0.5 nm, helps preserve assay performance. In addition, implementing double-referencing strategies effectively reduces non-specific interactions and improves the overall accuracy of kinetic measurements.
Ensure product purity: Discover our services for Impurity Profiling of Biosimilars.
Statistical Frameworks and the Quality Range Approach
Statistical assessment of analytical similarity relies on quantitative acceptance ranges established from multiple batches of the reference product to determine acceptable similarity margins. Although the traditional Quality Range (QR) methodology remains widely used, advanced analytical programs increasingly incorporate the Quality Range Maximum Likelihood (QRML) approach because it accounts for both within-batch and between-batch sources of variability.
For quantitative critical quality attributes (CQAs), statistical evaluation is generally organized into three distinct tiers based on the clinical significance of each attribute.
Tier 1 (Equivalence Testing)
Tier 1 is reserved for attributes of the highest clinical importance, including target-binding potency and primary structural variants. This approach evaluates whether the 90% confidence interval of the biosimilar mean remains within a predefined equivalence margin established for the reference product.
Tier 2 (Quality Range Approach)
Tier 2 applies to attributes with moderate clinical significance. In this approach, the acceptable similarity range is determined using the mean and standard deviation of the reference product according to the following equation:
Quality Range = μref ± k × SDref
where k represents a predefined multiplier, typically 1.5, 2, or 3, selected based on the clinical relevance and risk associated with the specific quality attribute.
Tier 3 (Descriptive Comparison)
Tier 3 is used for lower-criticality or qualitative attributes and primarily relies on descriptive statistical analyses together with graphical or visual comparisons to assess similarity.

Standard Quality Range vs. Quality Range Maximum Likelihood (QRML)
Although the conventional Quality Range (QR) method remains an accepted statistical approach for biosimilar analytical comparisons, it has several recognized limitations. Standard QR calculations typically rely on a single analytical measurement for each reference lot, causing the calculated acceptance limits to become highly variable when only a limited number of batches are available. Furthermore, the traditional QR approach does not adequately account for lot-to-lot manufacturing variability, shifts in batch means, or differences in variability between the reference product and the biosimilar candidate.
To overcome these shortcomings, advanced analytical programs increasingly implement the Quality Range Maximum Likelihood (QRML) methodology. QRML employs a two-level nested linear statistical model that independently estimates both within-lot variance, representing analytical method uncertainty, and between-lot variance, representing manufacturing process variability associated with the reference product. The standard deviation used to establish similarity limits is calculated as:
σQRML = √(σbetween2 + σwithin2)
By incorporating both analytical and manufacturing sources of variability, the QRML approach generates statistically robust and scientifically defensible acceptance limits. This methodology reduces the probability of incorrectly rejecting a truly biosimilar product while strengthening the overall reliability of analytical comparability assessments.
Navigating Patent and Reference Product Exclusivity Windows
Careful assessment of the 12-year reference product exclusivity period, together with evaluation of the active patent portfolio, is essential for determining the earliest legally permissible market entry for a biosimilar. Developers should cross-reference exclusivity timelines published in the FDA’s Purple Book while simultaneously conducting comprehensive patent landscape analyses to minimize litigation risks and develop effective commercialization strategies.
The Reference Product Exclusivity (RPE) period operates independently of the patent protection associated with the reference biologic. Under the Biologics Price Competition and Innovation Act (BPCIA), an approved innovator biologic is granted 12 years of regulatory exclusivity beginning on the date of its initial licensure. In addition, a separate 4-year restriction prevents the FDA from accepting a 351(k) biosimilar application during the initial post-approval period, creating a clearly defined regulatory barrier for market entry.
Even if all patents protecting the innovator biologic expire before the exclusivity period ends, the Reference Product Exclusivity (RPE) continues to prohibit FDA approval of competing biosimilars. Conversely, patent protection may extend well beyond the 12-year exclusivity period through secondary patents that cover formulations, manufacturing processes, delivery technologies, or specific therapeutic indications.
Conclusion
Successfully developing a commercially competitive biosimilar requires an integrated development strategy that combines high-resolution analytical characterization, rigorous statistical evaluation, and scientifically justified reference product sourcing practices for Biosimilar Comparability Studies. Continued harmonization of international regulatory requirements, particularly the reduced emphasis on comparative clinical efficacy studies, has established structural and functional analytical similarity as the primary evidence supporting biosimilarity.
By incorporating advanced mass spectrometry, orthogonal aggregate characterization techniques, and statistically robust methodologies such as the Quality Range Maximum Likelihood (QRML) approach, developers can effectively characterize the natural variability of the originator biologic while minimizing clinical development uncertainty and regulatory risk.
ResolveMass Laboratories Inc. serves as a trusted analytical partner by providing high-resolution mass spectrometry, advanced polymer characterization, and GLP/GMP-compliant analytical data packages designed to support successful biosimilar regulatory submissions across global markets.
For Expert Guidance and Customized Analytical Development
Frequently Asked Questions
The FDA’s March 2026 Revision 4 guidance removed the routine requirement for a three-way pharmacokinetic (PK) bridging study when a non-U.S.-licensed comparator is used. Instead, developers can support the use of a foreign comparator through comprehensive analytical evidence and strong scientific justification. This regulatory change reduces unnecessary clinical studies, shortens development timelines, and significantly lowers overall development costs. In many cases, sponsors may reduce PK study expenses by nearly half.
No. Regulatory agencies require that biosimilar candidates be compared only with the original reference biologic that was approved using a complete quality, non-clinical, and clinical data package. Approved biosimilars cannot be used as reference products because doing so could introduce cumulative analytical differences over successive generations of products. Using the original innovator biologic helps maintain scientific consistency and protects product quality, safety, and efficacy.
Modern analytical technologies can detect structural and functional differences between biologics with much greater sensitivity than traditional clinical efficacy studies. Extensive regulatory experience has shown that comparative clinical trials rarely identify differences that are not already evident through analytical characterization or pharmacokinetic evaluations. As a result, many regulatory agencies now prioritize comprehensive analytical similarity, in vitro functional testing, and PK/PD studies. This evidence-based approach improves efficiency without compromising patient safety.
The standard Quality Range (QR) approach establishes similarity limits using measurements from individual reference product lots, but its results can become unreliable when only a limited number of batches are available. Quality Range Maximum Likelihood (QRML) improves upon this method by separating analytical variability from manufacturing variability through a nested statistical model. This provides more representative acceptance limits that better reflect the natural variability of the reference product. Consequently, QRML offers stronger statistical confidence and more scientifically defensible similarity assessments.
Although earlier development programs often evaluated only a few reference batches, current best practices recommend analyzing approximately 10 to 15 independent lots. Assessing multiple lots produced at different manufacturing dates and various stages of shelf life provides a more accurate representation of the originator product’s natural variability. This broader dataset helps establish realistic analytical acceptance ranges and reduces the likelihood of creating overly restrictive product specifications during commercial manufacturing.
A comprehensive characterization strategy for monoclonal antibodies includes evaluating interactions with the target antigen, the neonatal Fc receptor (FcRn), complement protein C1q, and the complete Fcγ receptor family. These assays measure binding affinity, specificity, and kinetic behavior to confirm that the biosimilar performs similarly to the reference product. Together, they verify Fc-mediated effector functions, immune interactions, and mechanisms that influence antibody clearance. Such functional assessments are essential for demonstrating biosimilarity.
The neonatal Fc receptor (FcRn) plays a vital role in extending the circulation time of IgG antibodies by protecting them from intracellular degradation. It binds antibodies within acidic endosomes and releases them back into the bloodstream at physiological pH, allowing them to be recycled instead of destroyed. Even small changes in FcRn binding behavior can influence antibody clearance and serum half-life. Therefore, evaluating FcRn binding kinetics is an important component of biosimilar functional characterization.
The FDA’s Purple Book focuses on biological products, whereas the Orange Book primarily covers small-molecule drugs. The Purple Book provides information about licensed biologics, biosimilar approvals, interchangeable products, reference product exclusivity periods, and first licensure dates. Historically, it contained less comprehensive patent information than the Orange Book, although recent regulatory updates have expanded patent disclosure requirements. Developers commonly use the Purple Book when planning biosimilar development and market entry strategies.
Reference:
- Vulto, A. G., et al. (2025). Toward regulatory convergence and streamlined biosimilar development: Recommendations from an international qualitative study. BioDrugs. Advance online publication. https://doi.org/10.1007/s40259-025-00739-z
- Marini, J. C., Anderson, M., Cai, X.-Y., Chappell, J., Coffey, T., Gouty, D., Kasinath, A., Koppenburg, V., Oldfield, P., Rebarchak, S., & Bowsher, R. R. (2014). Systematic verification of bioanalytical similarity between a biosimilar and a reference biotherapeutic: Committee recommendations for the development and validation of a single ligand-binding assay to support pharmacokinetic assessments. The AAPS Journal, 16(6), 1149–1158. https://doi.org/10.1208/s12248-014-9669-5
- Health Canada. (2026, May 21). Summary of changes: Guidance on information and submission requirements for biosimilar biologic drugs. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/information-submission-requirements-biosimilar-biologic-drugs/summary-changes.html
- European Medicines Agency. (n.d.). Biosimilar medicines: Marketing authorisation. https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/biosimilar-medicines-marketing-authorisation
- Health Canada. (2025, December 18). Guidance on information and submission requirements for biosimilar biologic drugs. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/information-submission-requirements-biosimilar-biologic-drugs.html
- Kirchhoff, C. F., Wang, X.-Z. M., Conlon, H. D., Anderson, S., Ryan, A. M., & Bose, A. (2017). Biosimilars: Key regulatory considerations and similarity assessment tools. Biotechnology and Bioengineering, 114(12), 2696–2705. https://doi.org/10.1002/bit.26438

