
Introduction:
Bioequivalence study bioanalytical services are the laboratory backbone of every generic drug approval, formulation change, and lifecycle management submission that relies on comparative pharmacokinetics. Without a validated, regulator-ready bioanalytical method, a bioequivalence (BE) study cannot produce data that Health Canada, the FDA, or the EMA will accept — no matter how well the clinical portion of the trial was executed. For sponsors building an Abbreviated New Drug Submission (ANDS), ANDA, or EU generic dossier, the bioanalytical laboratory is often the single point of failure that determines whether years of formulation work translate into an approved product.
Many sponsors manage this risk by working with a partner offering regulated bioanalytical services for NDA and BLA submission alongside dedicated GLP bioanalytical services, rather than treating BE bioanalytical work as a standalone, one-off engagement.
This article walks through what regulators actually require from bioanalytical methods supporting BE studies, and what to look for when selecting a contract research organization (CRO) to run that work.
Summary:
- Bioequivalence study bioanalytical services generate the validated pharmacokinetic (PK) concentration data — typically LC-MS/MS-based — that regulators use to confirm a generic or modified formulation performs like the reference product.
- Health Canada, the FDA, and the EMA all expect method validation aligned with ICH M10, though submission formats and acceptance criteria differ by region.
- Core validation parameters include selectivity, sensitivity (LLOQ), accuracy, precision, matrix effect, recovery, and stability across freeze-thaw, bench-top, and long-term storage conditions.
- Choosing a bioanalytical CRO comes down to five factors: regulatory inspection history, method development depth, matrix and analyte experience, data integrity infrastructure, and turnaround reliability.
- ResolveMass Laboratories supports bioequivalence sponsors with ICH M10-aligned bioanalytical method development, validation, and sample analysis backed by a Canadian GMP/GLP-oriented quality system.
1: What Are Bioequivalence Study Bioanalytical Services?
Bioequivalence study bioanalytical services refer to the method development, validation, and sample analysis work used to measure drug (and metabolite) concentrations in biological matrices — usually plasma, serum, or urine — collected from BE study subjects. The resulting concentration-time data feed directly into PK parameters like Cmax, AUC0-t, and AUC0-∞, which statisticians then use to determine whether a test product is bioequivalent to its reference.
In practice, this work spans three linked stages:
- Method development — building a selective, sensitive assay (typically LC-MS/MS) for the parent drug and relevant metabolites, drawing on dedicated LC-MS/MS bioanalytical services capability
- Method validation — formally demonstrating the assay meets regulatory acceptance criteria
- Study sample analysis — running actual subject samples under the validated method, with incurred sample reanalysis (ISR) as a confirmatory check
For modalities beyond small molecules, this same three-stage approach extends to peptide bioanalytical services and biomarker bioanalytical services, which often require distinct extraction chemistry and assay formats compared to conventional small-molecule LC-MS/MS work.
2: Why Do Regulators Scrutinize Bioanalytical Data So Closely?
Regulators closely scrutinize bioanalytical data because the validity of a bioequivalence (BE) study depends entirely on the accuracy and reliability of the measured drug concentrations. Pharmacokinetic (PK) parameters such as Cmax, AUC, and Tmax are calculated from these concentration data, meaning any analytical error can directly affect the study’s conclusions. An inaccurate or poorly validated bioanalytical method could make a non-equivalent product appear bioequivalent—or incorrectly suggest that an equivalent product has failed.
Since regulatory approval for generic medicines relies heavily on statistical comparisons of PK parameters, even small systematic errors in bioanalysis can lead to incorrect regulatory decisions. For this reason, agencies such as the FDA, EMA, and Health Canada require bioanalytical methods to undergo rigorous validation for accuracy, precision, selectivity, sensitivity, recovery, matrix effects, stability, and reproducibility before they are used in pivotal studies.
As a result, the bioanalytical section is one of the most thoroughly reviewed components of a bioequivalence submission. Regulatory inspectors also conduct routine and for-cause Good Laboratory Practice (GLP) inspections of bioanalytical Contract Research Organizations (CROs) to verify compliance with quality systems, data integrity requirements, and validated analytical procedures. Robust bioanalytical data not only strengthen regulatory confidence but also reduce the risk of study rejection, repeat trials, and costly development delays.
3: What Regulatory Requirements Apply to Bioanalytical Methods for BE Studies?
Bioanalytical methods supporting BE studies must be validated according to ICH M10 principles, with region-specific expectations layered on top depending on whether the submission goes to Health Canada, the FDA, or the EMA. ICH M10, finalized in 2022, harmonized much of what were previously separate FDA and EMA bioanalytical method validation guidances, though agencies retain some jurisdiction-specific expectations during review. This is the same validation rigor that underpins broader bioanalytical method development and validation work across other regulated submission types, not just BE studies specifically.
Core Validation Parameters
| Parameter | What It Demonstrates | Typical Acceptance Criteria |
|---|---|---|
| Selectivity | No interference from matrix components or co-administered drugs | Response <20% of LLOQ (<5% for internal standard) |
| Sensitivity (LLOQ) | Lowest concentration reliably quantified | Signal-to-noise ≥5; accuracy/precision within ±20% |
| Accuracy & Precision | Assay measures true concentration consistently | Within ±15% of nominal (±20% at LLOQ) |
| Matrix Effect | Ion suppression/enhancement from biological matrix | CV ≤15% across matrix lots |
| Recovery | Extraction efficiency of analyte | Consistent and reproducible across QC levels |
| Stability | Analyte integrity through handling, storage, freeze-thaw | Within ±15% of initial concentration |
Region-Specific Considerations
- Health Canada generally follows ICH M10 but retains specific documentation expectations for ANDS bioanalytical reports, and Canadian GLP compliance is assessed against Health Canada’s own inspection program. This dual expectation is why sponsors often look for biomarker bioanalytical services for FDA and Health Canada that are built around both agencies from the outset.
- U.S. FDA requires bioanalytical data to support ANDA submissions under 21 CFR Part 320, with incurred sample reanalysis and cross-validation data expected when methods or sites change mid-study. Sponsors filing in the U.S. specifically should confirm their CRO’s approach to bioanalytical method development in the United States matches current FDA expectations.
- EMA applies ICH M10 alongside its own guideline on bioanalytical method validation, with particular attention to partial validation and cross-validation when multiple laboratories or methods are involved in one study.
Sponsors running multi-region filings should confirm their bioanalytical CRO can produce a single validation package that satisfies all target agencies, rather than running duplicate validations per jurisdiction. A CRO offering bioanalytical services in North America with a unified quality system is typically better positioned to support this than one coordinating across multiple disconnected sites.
4: What Should Sponsors Look for When Selecting a Bioanalytical CRO?
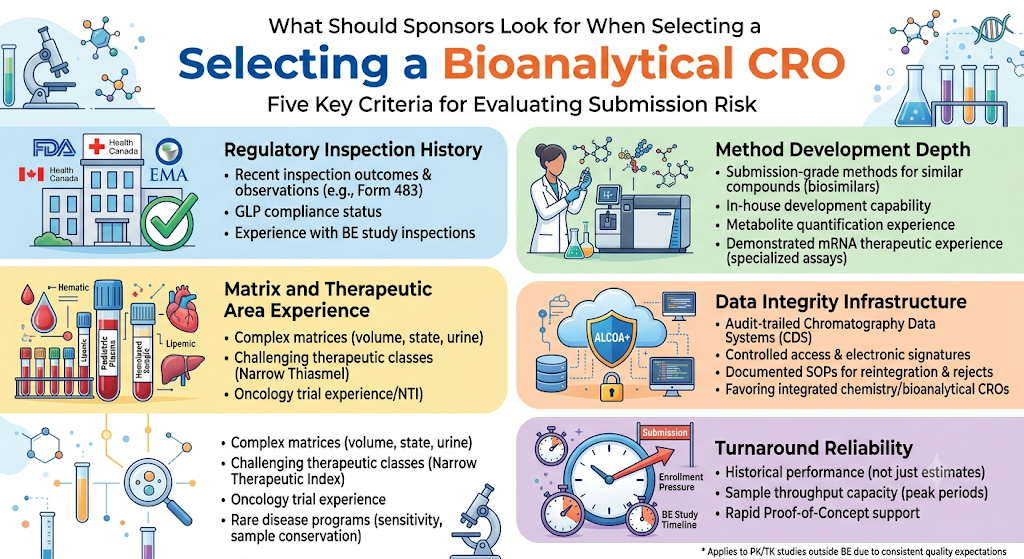
Sponsors should evaluate a bioanalytical CRO on five criteria: demonstrated regulatory inspection history, method development depth for the specific analyte class, relevant matrix and therapeutic area experience, data integrity infrastructure, and realistic turnaround commitments. Each of these directly affects submission risk, not just study cost or timeline. Many of these same criteria apply equally when evaluating bioanalytical CRO services for PK and TK studies outside the BE context, since the underlying quality expectations are consistent.
1. Regulatory Inspection History
A CRO’s track record with Health Canada, FDA, or EMA inspections is the clearest predictor of how its data will hold up under review. Ask for:
- Recent inspection outcomes and any Form 483 or equivalent observations
- GLP compliance status and certificate scope
- Experience specifically with BE study inspections, not just general bioanalytical work
2. Method Development Depth
Not every CRO has equal depth across analyte classes. A lab strong in small-molecule LC-MS/MS may have limited experience with peptides, biologics, or complex prodrugs requiring specialized extraction chemistry. Confirm the CRO has:
- Prior published or submission-grade methods for structurally similar compounds, including LC-MS/MS services for biosimilars where relevant
- In-house method development capability rather than reliance on published literature methods alone
- Experience with metabolite quantification when active metabolites are relevant to the BE assessment
- For newer modalities, demonstrated bioanalytical method development for mRNA therapeutics or comparable nucleic-acid-based assay experience, since these require validation approaches distinct from conventional small-molecule methods
3. Matrix and Therapeutic Area Experience
Complex matrices (pediatric plasma volumes, hemolyzed or lipemic samples, urine for renally cleared drugs) and challenging therapeutic classes (narrow therapeutic index drugs, highly protein-bound compounds) benefit from a CRO with direct prior experience, since these scenarios often require non-standard validation approaches. This is especially relevant for sponsors seeking a bioanalytical CRO for oncology clinical trials, where narrow therapeutic windows are common, or for bioanalytical services for rare disease programs, where limited sample volumes and small patient populations demand extra assay sensitivity and conservation of material.
4. Data Integrity Infrastructure
Regulators increasingly scrutinize data integrity (ALCOA+ principles) as closely as the science itself. This is one reason sponsors favor an integrated chemistry and bioanalytical CRO, where data systems and quality oversight are unified across services rather than fragmented across vendors. Look for:
- Audit-trailed chromatography data systems (CDS)
- Controlled access and electronic signature workflows
- Documented SOPs for reintegration, reprocessing, and reason-for-reject records
5. Turnaround Reliability
BE study timelines are frequently compressed by clinical enrollment pressure. A CRO’s stated turnaround should be evaluated against its actual historical performance, not just proposal-stage estimates, since bioanalytical delays are one of the most common causes of BE study timeline slippage. Ask specifically about bioanalytical sample throughput capacity during peak enrollment periods, and whether the CRO can support bioanalytical services for rapid proof-of-concept work if early feasibility data is needed before committing to full study volumes.
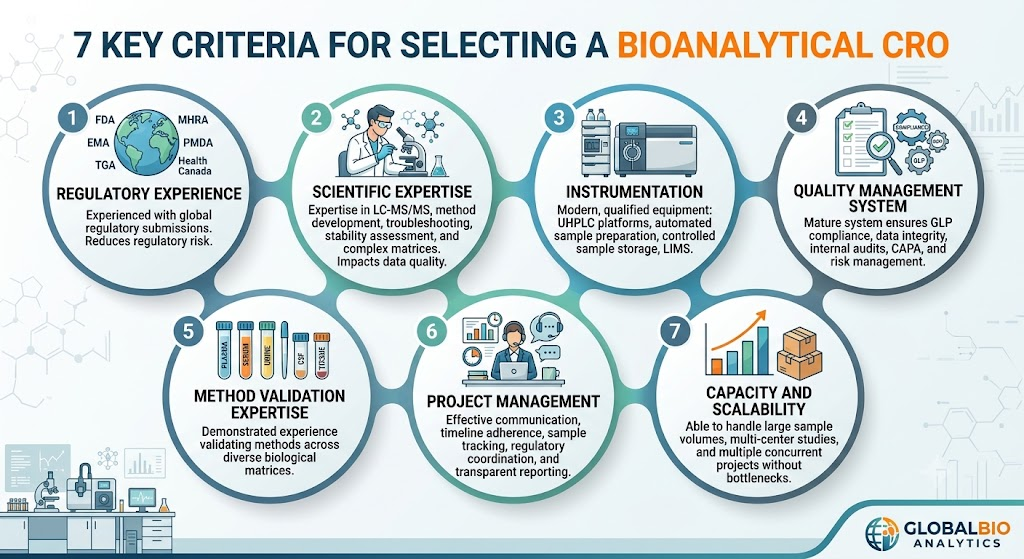
5: What Should You Look for When Selecting a Bioanalytical CRO?
The ideal CRO combines scientific expertise, regulatory experience, robust quality systems, and advanced analytical capabilities.
Below are the most important evaluation criteria.
1. Regulatory Experience
Choose laboratories experienced with submissions to:
- US FDA
- EMA
- Health Canada
- MHRA
- PMDA
- TGA
Experience with multiple agencies reduces regulatory risk.
2. Scientific Expertise
Experienced scientists should possess expertise in:
- LC-MS/MS
- Method development
- Troubleshooting
- Matrix effect evaluation
- Complex biological matrices
- Stability assessment
Scientific knowledge directly impacts data quality.
3. Instrumentation
Modern laboratories should operate:
- Triple quadrupole LC-MS/MS systems
- UHPLC platforms
- Automated sample preparation
- Temperature-controlled sample storage
- Qualified laboratory information systems
Advanced equipment improves reproducibility and turnaround time.
4. Quality Management System
Evaluate whether the CRO maintains:
- GLP compliance
- SOP-driven operations
- Electronic data integrity
- Audit trails
- Internal quality audits
- CAPA system
- Risk management program
A mature quality system supports consistent regulatory compliance.
5. Bioanalytical Method Validation Expertise
Look for demonstrated experience in validating methods for:
- Plasma
- Serum
- Whole blood
- Urine
- Tissue homogenates
- Cerebrospinal fluid
Different biological matrices require specialized analytical expertise.
6. Project Management
Effective project management ensures:
- Clear communication
- Timeline adherence
- Sample tracking
- Regulatory coordination
- Issue resolution
- Transparent reporting
Dedicated project managers improve study efficiency.
7. Capacity and Scalability
An experienced CRO should comfortably handle:
- Pilot studies
- Pivotal bioequivalence studies
- Large sample volumes
- Multi-center clinical studies
- Multiple concurrent projects
Scalable operations prevent analytical bottlenecks.
6: How Does ResolveMass Support Bioequivalence Bioanalytical Programs?
ResolveMass Laboratories supports bioequivalence sponsors through ICH M10-aligned bioanalytical method development, full validation, and study sample analysis for small-molecule and peptide analytes in plasma, serum, and urine matrices, delivered as part of a broader suite of bioanalytical services. As a Canadian analytical CRO with mass spectrometry-focused characterization expertise, ResolveMass applies the same rigor used in its extractables and leachables, nitrosamine, and peptide characterization work to bioanalytical assay development — including LC-MS/MS method optimization, matrix-specific extraction development, and stability assessments designed to withstand regulatory scrutiny.
For sponsors weighing internal capability against external support, ResolveMass also offers cost-effective bioanalytical services structured around bioanalytical outsourcing and outsourced bioanalytical services models, including dedicated biomarker bioanalytical services CRO support for programs that require biomarker data alongside standard PK endpoints. As emerging discovery approaches reshape early development, ResolveMass also supports sponsors exploring a bioanalytical CRO for AI drug discovery pipeline, ensuring bioanalytical strategy is aligned from candidate selection through to BE study execution.
Conclusion:
Reliable bioequivalence study bioanalytical services are not a commodity lab service — they are a regulatory risk decision. A method that is technically functional but poorly documented, or a CRO with a thin inspection history, can jeopardize an otherwise sound BE study. Sponsors should evaluate bioanalytical partners against ICH M10 alignment, region-specific regulatory expectations, and the five CRO selection criteria outlined above before committing study samples to any laboratory.
For sponsors evaluating a bioanalytical partner for an upcoming BE program, the ResolveMass team is available to discuss method feasibility and validation scope for specific analytes and matrices.
Frequently Asked Questions:
Bioequivalence Study Bioanalytical Services provide the scientific evidence needed to demonstrate that a generic drug is bioequivalent to the innovator product. Regulatory agencies require precise measurement of drug concentrations in biological samples to compare the absorption and exposure of both products. High-quality bioanalytical data reduce regulatory concerns and improve the chances of successful approval. Poor analytical performance can invalidate an entire bioequivalence study, resulting in delays and additional costs. Reliable bioanalysis is therefore a critical component of the generic drug development process.
Bioanalytical testing measures the concentration of a drug and, when applicable, its metabolites in biological samples collected from study participants. These concentration values are used to construct pharmacokinetic profiles and calculate parameters such as Cmax, Tmax, and AUC. The resulting data help determine whether the test and reference products have equivalent bioavailability. Accurate testing is essential for ensuring that pharmacokinetic comparisons are scientifically valid. Regulatory agencies rely on these results when evaluating bioequivalence submissions.
Plasma is the most commonly analyzed biological matrix in bioequivalence studies because it reflects systemic drug exposure. Depending on the drug and study design, other matrices such as serum, whole blood, urine, saliva, cerebrospinal fluid, or tissue samples may also be analyzed. The choice of biological matrix depends on the pharmacokinetic characteristics of the drug and regulatory expectations. Each matrix requires specific validation to ensure analytical accuracy and precision. Proper sample collection, storage, and handling are essential for maintaining data quality.
Regulatory agencies require bioanalytical methods to be validated before they are used for clinical sample analysis. Validation typically includes assessments of accuracy, precision, selectivity, sensitivity, calibration curve performance, recovery, matrix effects, carryover, dilution integrity, and analyte stability. Documentation must demonstrate that the method consistently produces reliable and reproducible results. Validation studies should follow applicable regulatory guidance and Good Laboratory Practice (GLP) principles. A well-validated method reduces the likelihood of regulatory questions during submission review.
Several regulatory authorities publish guidance for bioanalytical method validation and bioequivalence studies. These include the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), Health Canada, the Medicines and Healthcare products Regulatory Agency (MHRA), and other national agencies. Although individual requirements may vary slightly, they generally emphasize validated methods, data integrity, quality systems, and comprehensive documentation. Following internationally recognized guidance helps ensure that bioanalytical data are accepted across multiple regulatory jurisdictions.
A bioequivalence study may fail due to poor study design, inadequate analytical method validation, high variability in drug concentrations, improper sample handling, or protocol deviations. Analytical issues such as matrix effects, carryover, instability of the analyte, or instrument performance problems can also compromise data quality. In addition, failure to comply with regulatory guidelines or quality system requirements may result in rejected data. Working with an experienced bioanalytical CRO helps identify and mitigate these risks before they affect the study outcome. Consistent quality control and robust analytical procedures are essential for successful bioequivalence studies.
Get In Touch With Us
Reference
- Boterman M, Doig M, Breda M, Lowes S, Jersey J, Shoup R, Garofolo F, Dumont I, Martinez S, Needham S, Caturla MC. Recommendations on the interpretation of the new European Medicines Agency guideline on bioanalytical method validation by Global CRO Council for Bioanalysis (GCC). Bioanalysis. 2012 Mar 1;4(6):651-60.https://www.tandfonline.com/doi/abs/10.4155/bio.12.18
- Desai S, Patel N. Checklist to select contract Research Organization for early phase Bioavailability/Bioequivalence Clinical Studies in Healthy Adult Human Volunteers. Research Journal of Pharmacology and Pharmacodynamics. 2021 Oct;13(4):131-2.https://indianjournals.com/api/article-view/rjppd-13-4-005
- Lowes S, Jersey J, Shoup R, Garofolo F, Needham S, Couerbe P, Lansing T, Bhatti M, Sheldon C, Hayes R, Islam R. Conference report: 4th Global CRO Council for Bioanalysis: coadministered drugs stability, EMA/US FDA Guidelines, 483s and carryover.https://www.tandfonline.com/doi/abs/10.4155/bio.12.48
- Noonan PK. Outsourcing Bioavailability and Bioequivalence Studies to Contract Research Organizations. InGeneric Drug Product Development 2014 Oct 31 (pp. 321-364). CRC Press.https://www.taylorfrancis.com/chapters/edit/10.1201/9781420030419-16/outsourcing-bioavailability-bioequivalence-studies-contract-research-organizations-patrick-noonan
- Noonan PK. Bioavailability and Bioequivalence Studies to Contract Research Organizations. Generic Drug Product Development: Solid Oral Dosage Forms. 2013 Oct 24:265.https://books.google.com/books?hl=en&lr=&id=YfVCEQAAQBAJ&oi=fnd&pg=PA265&dq=Bioequivalence+Study+Bioanalytical+Services:+Regulatory+Requirements+and+CRO+Selection+Criteria&ots=c1PTUYI6DJ&sig=VEr3JY5fXNwXhxnpJ809Yf_fYEw

