
The assessment of Extractables and Leachables (E&L) in biologic drug products remains one of the most significant safety and quality concerns in biopharmaceutical development. Complex therapeutic protein formulations are particularly susceptible to trace-level chemical contaminants that can migrate into the product. Since biologics are manufactured using living host cell systems and are typically formulated in aqueous environments containing carefully controlled pH conditions and surfactants, they are especially vulnerable to interactions with chemical species originating from container closure systems and manufacturing equipment. This technical report examines advanced mass spectrometry technologies, applicable regulatory frameworks, and analytical threshold calculations necessary for the effective control of extractables and leachables in biologic therapeutic products.
For a detailed overview of the potential risks and industry standards, explore Extractables and Leachables in Pharmaceutical Products.
Share via:
Article Summary:
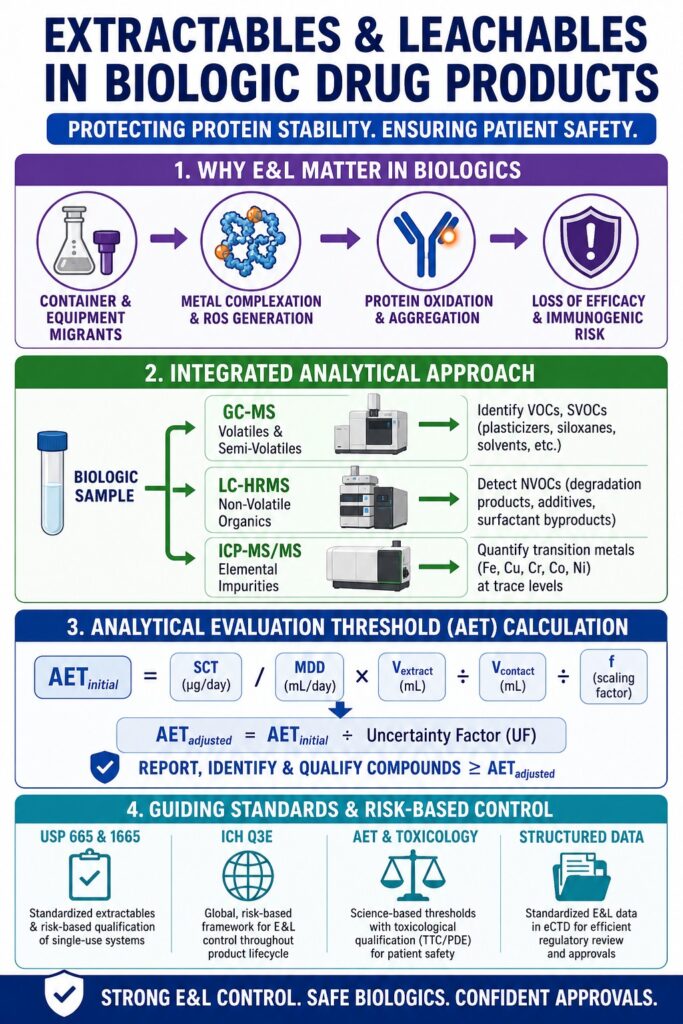
- Extractables and Leachables (E&L) are chemical substances that can migrate from packaging materials, manufacturing equipment, and single-use systems into biologic drug products, potentially affecting product quality and patient safety.
- Biologics such as monoclonal antibodies, fusion proteins, and antibody-drug conjugates are particularly vulnerable to trace contaminants because their therapeutic activity depends on maintaining complex protein structures.
- Metal ions and other leachables can trigger oxidative stress, protein degradation, and aggregation, which may reduce drug effectiveness and increase the risk of unwanted immune responses, including anti-drug antibody formation.
- Comprehensive E&L characterization requires multiple analytical techniques, including GC-MS for volatile compounds, LC-HRMS for non-volatile organic contaminants, and ICP-MS for elemental impurities and trace metals.
- The Analytical Evaluation Threshold (AET) converts toxicological safety limits into measurable laboratory reporting thresholds, helping scientists identify compounds that require further toxicological assessment.
- Regulatory frameworks such as USP 665, USP 1665, and the emerging ICH Q3E guideline promote a risk-based approach for evaluating materials, qualifying single-use systems, and controlling E&L risks throughout the product lifecycle.
- Structured E&L data, supported by robust analytical testing and traceability between extractables and leachables, plays a critical role in regulatory submissions, helping manufacturers demonstrate product safety, maintain compliance, and avoid approval delays.
How Do Extractables and Leachables in Biologic Formulations Threaten Protein Stability?
Chemical contaminants released from packaging components or bioprocessing equipment can directly affect the quality of biologic products by promoting protein oxidation, inducing aggregation, and modifying post-translational structures. These interactions may reduce therapeutic effectiveness while substantially increasing the potential for immunogenic reactions in patients.
Unlike small-molecule drugs, which are chemically synthesized and generally possess stable molecular structures, biopharmaceutical products such as monoclonal antibodies (mAbs), fusion proteins, and antibody-drug conjugates (ADCs) depend on delicate non-covalent interactions to preserve their biologically active higher-order structures. Even trace concentrations of contaminants within the formulation can disrupt these interactions, resulting in both chemical and physical degradation. One of the most important mechanisms responsible for chemical degradation is metal-catalyzed oxidation (MCO). Transition metals, including iron (Fe³⁺/Fe²⁺), copper (Cu²⁺/Cu⁺), chromium, cobalt, and nickel, are recognized leachables that may originate from stainless steel bioreactors, glass containers, and elastomeric components such as syringe plungers and vial stoppers.
Proteins frequently function as complexing agents for transition metals. This interaction can inhibit the development of the protective oxide passivation layer on stainless steel surfaces, thereby accelerating the release of metal ions into the manufacturing stream. Once present within the formulation, these transition metals undergo redox cycling. In the presence of electron donors, they generate highly reactive oxygen species (ROS), including the hydroxyl radical (•OH), through Fenton-like reactions. These ROS preferentially attack susceptible amino acid residues, particularly methionine, cysteine, histidine, tryptophan, and tyrosine.
[ Transition Metal Leaching ] (Fe, Cu, Cr, Co, Ni)
│
▼
[ Complexation with Active Protein ]
│
▼
[ ROS Generation (Fenton/Haber-Weiss) ]
│
▼
[ Oxidation of Susceptible Amino Acids (Met, Cys, His) ]
│
▼
[ Hydrophobic Exposure ──► Irreversible Aggregation ]Oxidation of specific residues, including Methionine 255 and Methionine 431 located within the heavy chain of an IgG1 monoclonal antibody, can produce conformational changes in the protein structure. These alterations expose hydrophobic regions that are normally buried within the protein core, initiating self-association processes that eventually lead to irreversible soluble or insoluble aggregation. Such aggregation introduces substantial clinical concerns, including the formation of anti-drug antibodies (ADAs), which may neutralize therapeutic activity or provoke systemic hypersensitivity reactions.
Biologic formulations commonly contain polysorbate surfactants, such as polysorbate 80, to minimize surface-induced aggregation. However, these surfactants may undergo auto-oxidation, generating reactive fatty acid peroxides capable of degrading both proteins and polymeric packaging materials. This oxidative process may also enhance the migration of organic additives, including 2-ethylhexanoic acid (2-EHA), as well as rubber vulcanization agents such as 2-mercaptobenzothiazole (MBT).
In advanced therapeutic modalities, including cell and gene therapy (CGT) products, process equipment-related leachables (PERLs) may interact directly with living cellular systems. Leachables released from polymer-based microcarriers, transfer tubing, and cryopreservation bags have demonstrated adverse effects on cell viability, metabolic performance, and growth characteristics in Chinese Hamster Ovary (CHO) cells, CAR T cells, and human mesenchymal stem cells (MSCs).
What Analytical Platforms are Mandatory to Resolve Extractables and Leachables in Biologic Matrices?
Comprehensive characterization of potential chemical migrants in complex biopharmaceutical formulations requires an integrated analytical strategy combining gas chromatography, liquid chromatography, and inductively coupled plasma mass spectrometry. Since no single analytical technique can adequately detect volatile, semi-volatile, non-volatile, and inorganic contaminants simultaneously, regulatory agencies require an orthogonal analytical approach.
To learn more about selecting the right methodologies, see GC-MS vs LC-MS in Extractables and Leachables Testing.
One of the primary analytical challenges associated with biologic formulations is the complexity of the sample matrix. Elevated protein concentrations, viscous excipients, and active surfactants contribute significant spectral and ionization interference. Direct injection of biologic drug products into high-resolution mass spectrometers can result in column contamination, increased backpressure, and severe ion suppression, thereby obscuring low-level chemical migrants.
Consequently, advanced sample preparation methods are essential. These methods may include targeted protein precipitation using organic solvents such as chilled acetonitrile, solid-phase extraction (SPE) employing mixed-mode sorbents, or liquid-liquid extraction (LLE) specifically designed to separate organic contaminants from hydrophilic protein matrices. Analytical workflows should also include appropriate control blanks to distinguish background contaminants from genuine container closure or process-related migrants.
For volatile organic compounds (VOCs) and low-molecular-weight monomers, Headspace Gas Chromatography-Mass Spectrometry (HS-GC-MS) remains the preferred analytical platform. HS-GC-MS operates by heating samples to volatilize analytes into the headspace, thereby minimizing matrix-related interferences. Semi-volatile organic compounds (SVOCs), including plasticizers, phthalates, antioxidants, and siloxanes, are generally analyzed using Direct Injection GC-MS operating under Electron Ionization (EI) conditions.
Electron ionization generates highly reproducible fragmentation patterns that can be compared against established NIST and Wiley spectral libraries for rapid compound identification. Nevertheless, polar or thermally labile SVOCs frequently require chemical derivatization prior to GC-MS analysis to achieve suitable chromatographic performance.
For polar, non-volatile organic compounds (NVOCs), including polymer degradation products, photoinitiators, and surfactant degradation byproducts, Liquid Chromatography coupled with High-Resolution Mass Spectrometry (LC-HRMS) is essential. Instrument platforms such as Quadrupole Time-of-Flight (QTOF) and Orbitrap mass spectrometers provide the resolution and mass accuracy required for comprehensive characterization. Operating LC-HRMS using both Electrospray Ionization (ESI) and Atmospheric Pressure Chemical Ionization (APCI) maximizes analyte coverage across a broad polarity range.
[ BIOLOGIC SAMPLE ]
│
┌─────────────────────┴─────────────────────┐
▼ ▼
[ Organic Fraction ] [ Inorganic Fraction ]
│ │
┌─────────┴─────────┐ ▼
▼ ▼ [ ICP-MS ]
[ GC-MS ] [ LC-MS ] (Transition Metals,
(Volatiles & (Non-Volatile Elemental Impurities)
Semi-Volatiles) Organics)Elemental impurities and trace transition metal contaminants are most effectively analyzed using Inductively Coupled Plasma Mass Spectrometry (ICP-MS/MS). This technique utilizes a collision/reaction cell, typically operated with helium or hydrogen gas, to remove polyatomic interferences and achieve sub-part-per-billion detection limits. Direct coupling of Size-Exclusion Chromatography (SEC) with ICP-MS/MS using PEEK capillary tubing enables metal-protein speciation studies, facilitating determination of whether transition metals are bound to active drug substances or remain present as free ions within the formulation.
For specific insights into metal analysis, refer to ICP-MS in Extractables and Leachables Testing.
How is the Analytical Evaluation Threshold Calculated to Ensure Safety for Extractables and Leachables in Biologic Products?
The Analytical Evaluation Threshold (AET) is determined by converting a clinically relevant safety threshold into an analytical concentration limit that accounts for the maximum daily dose and analytical uncertainty. This threshold establishes the reporting limit above which detected compounds must be identified and assessed toxicologically.
Understanding the calculation is vital; read more at AET for Extractables and Leachables Studies.
The mathematical basis of the AET serves as the connection between toxicological safety limits and laboratory analytical measurements. The initial AET (AETinitial), representing the concentration threshold within an extract or drug product, is calculated according to the following equation:
AETinitial = (SCT / MDD) × (Vextract / (Vcontact × f))
Where:
SCT is the Safety Concern Threshold (μg/day), representing the maximum acceptable daily intake below which a leachable is considered unlikely to present systemic toxicity or carcinogenic risk. For high-risk parenteral biotherapeutics, SCT is generally established at 1.5 μg/day. For genotoxic or mutagenic compounds, a more stringent limit of 0.15 μg/day is typically applied, corresponding to a lifetime cancer risk of 10⁻⁶.
MDD is the Maximum Daily Dose administered to the patient.
Vextract represents the volume of extraction solvent utilized during the controlled extraction study (mL).
Vcontact refers to the volume of drug product that directly contacts the manufacturing component or container closure system (mL).
f is a dimensionless packaging-to-product scaling factor used to adjust for differences between extraction conditions and actual dosage delivery conditions.
For direct drug product analyses where the final formulation itself is examined, the threshold may be simplified to a dose-based equation incorporating an Uncertainty Factor (UF):
AETadjusted = (SCT × Dose) / Uncertainty Factor
The inclusion of the Uncertainty Factor (UF) is necessary to account for variation in Relative Response Factors (RRFs). During non-targeted screening, unknown analyte concentrations are estimated by comparing chromatographic peak areas to those of a single internal standard. This approach assumes uniform detector responses across all analytes, an assumption that is rarely valid.
When an unknown compound possesses a low Relative Response Factor (RRF < 1), its concentration may be significantly underestimated. Consequently, the compound may appear to fall below the AET despite actually exceeding the safety threshold, resulting in a Type II false-negative outcome.
To compensate for this variability, the initial AET is adjusted downward through application of the Uncertainty Factor:
AETadjusted = AETinitial / UF
The Uncertainty Factor is calculated using the Relative Standard Deviation (RSD) of response factors obtained from a representative analyte database:
UF = 1 / (1 − RSD)
Historically, a default UF value of 2 was considered acceptable for gas chromatography applications. However, in LC-MS analyses, relative response factor variability can exceed 100% RSD.
An evaluation involving approximately 1,200 E&L compounds analyzed using GC-MS, LC-MS-APCI, and LC-MS-ESI demonstrated that a UF between 3 and 4 generally provides sufficient protection for GC-MS workflows, whereas LC-MS screening may require a UF as high as 10 to achieve approximately 97% detection coverage for low-response analytes.
To avoid generating excessive numbers of irrelevant analytical findings due to overly conservative AET values, many laboratories establish compound-specific RRF databases, commonly referred to as the “RRFlow” approach. This methodology mathematically normalizes peak responses, reducing detection bias while minimizing dependence on broad uncertainty factors.
| Parameter | Regulatory Context | High-Risk Parenteral | Ophthalmic Formulation | Orally Inhaled (OINDP) |
|---|---|---|---|---|
| SCT Limit | Safety threshold below which a leachable presents negligible safety risk. | 1.5 μg/day | Concentration-based threshold | 0.15 μg/day |
| MDD Range | Maximum daily administered dose. | 1 to 10 mL/day | 0.1 to 0.5 mL/day | 1 to 2 g/day |
| Uncertainty Factor | Safety factor derived from detector response variability. | UF = 4 to 10 (LC-MS) | UF = 5 (Standard) | UF = 2 to 5 (GC-MS) |
| AET Threshold | Reporting limit requiring structural identification. | 0.15 μg/mL | 1.0 ppm (Reporting) | 0.03 μg/device |
If you are concerned about project timelines, you can review factors affecting Cost of Extractables and Leachables Testing.
What Do USP 665 and USP 1665 Mandate for Single-Use Systems in Bioprocessing?
USP General Chapter 665 establishes standardized and enforceable requirements for extractables characterization of plastic materials used in biomanufacturing, while USP 1665 provides the risk-based framework used to qualify these systems for intended bioprocessing applications. Effective May 1, 2026, these standards require biopharmaceutical manufacturers to demonstrate that process equipment-related leachables do not adversely affect drug safety, identity, strength, quality, or purity.
Single-use systems (SUS), including disposable bioreactor bags, polymeric tubing, transfer connectors, process filters, and chromatography cassettes, have become widely adopted throughout bioprocessing because of their flexibility and operational efficiency. However, these materials often exhibit high surface-area-to-volume ratios, increasing the likelihood of additive migration, including slip agents, antioxidants, and plasticizers.
[ Step 1: Initial Material Contact Assessment ]
│
▼
[ Step 2: Establish Risk-Based Suitability ]
(Utilizing USP Matrix)
│
┌────────────────────┴────────────────────┐
▼ ▼
[ Low Risk (Score < 13) ] [ High Risk (Score ≥ 13) ]
│ │
▼ ▼
[ Minimal Characterization ] [ Comprehensive E&L Testing ]
(Baseline Supplier Data) (Standardized Multi-Solvent)USP 1665 utilizes a risk-scoring methodology based on several process-related factors:
Chemical Nature of the Process Stream: Materials contacting highly organic or surfactant-rich process fluids present elevated leaching risk, while sterile water and simple aqueous buffers typically pose lower risk.
Contact Temperature: Increasing processing or storage temperatures accelerate thermodynamically driven migration processes.
Contact Duration: Extended exposure periods, such as a 21-day cell culture operation, increase cumulative extractables loads compared to short-duration processes.
Material Construction and Reactivity: Polymeric materials containing multiple additives or exposed to gamma irradiation degradation present higher risk profiles.
Downstream Purification Steps: Components located upstream in the manufacturing process generally present lower final product risk because subsequent purification steps may remove migrated contaminants. In contrast, downstream components such as final filling needles and sterile filtration assemblies represent the highest risk because no additional clearance mechanisms exist prior to packaging.
For extractables characterization of polymeric materials, many organizations reference ASME BPE extraction protocols. These protocols include dynamic extraction conditions utilizing a surface-area-to-solvent ratio of 60 cm² per 20 mL of extraction fluid or a weight-to-volume ratio of 0.2 g per 1 mL.
Studies involving biopharmaceutical tubing materials, including silicone and Santoprene, have demonstrated the release of homologous silicone oligomers under dynamic flow conditions, along with plasticizers, phenolic compounds, and polymer antioxidants.
For deep dives into specific container types, see EL Testing for Pre-filled Syringes
and Extractables and Leachables Testing for Autoinjectors.
Importantly, biopharmaceutical manufacturers cannot consider supplier-provided extractables data packages as the final compliance requirement. Although suppliers characterize materials under standardized laboratory conditions, they cannot determine whether observed compounds at product-specific concentrations present patient safety concerns. The drug sponsor remains legally responsible for performing a product-specific leachables risk assessment and, when necessary, conducting direct leachables testing.
How Does the ICH Q3E Guideline Modernize the Control Strategy for Extractables and Leachables in Biologic Development?
The forthcoming ICH Q3E guideline introduces a globally harmonized, science-based, and risk-driven framework for evaluating and controlling extractable and leachable impurities throughout the product lifecycle. By integrating analytical screening with toxicological qualification, the guideline simplifies international regulatory submissions and replaces fragmented regional requirements.
Historically, E&L regulations were highly fragmented and relied on a combination of local standards, pharmacopeial requirements, and food-contact regulations. This lack of harmonization resulted in significant variability in study design and analytical expectations among global regulatory agencies.
The draft ICH Q3E guideline, developed by the International Council for Harmonisation, is intended to align with several established quality and safety guidelines, including:
- ICH Q3A and Q3B: Impurities in new drug substances and drug products.
- ICH Q3C: Residual solvents.
- ICH Q3D: Elemental impurities and Permitted Daily Exposure (PDE) limits.
- ICH M7: Mutagenic impurities and DNA-reactive structure assessment.
- ICH Q9: Quality Risk Management (QRM).
- ICH Q12: Lifecycle management and change control.
The guideline introduces a structured, risk-based framework incorporating material characterization, analytical evaluation thresholds, and potency-based classification systems for leachables.
Rather than imposing rigid testing requirements, the guideline emphasizes scientific justification and encourages collaboration between analytical chemists and toxicologists during development of an Analytical Target List (ATL).
Additionally, the USP is developing dosage form-specific chapters that complement these harmonization efforts. These chapters establish safety thresholds and testing expectations tailored to specific clinical applications.
For example, USP <1664.2> (Parenteral Drug Products) establishes an SCT of 1.5 μg/day, while USP <1664.3> (Topical Ophthalmic Drug Products) utilizes concentration-based limits, including a reporting threshold of 1 ppm, an identification threshold of 10 ppm, and a qualification threshold of 20 ppm, reflecting the unique toxicological considerations associated with ocular exposure.
Why is Structured E and L Data Essential for Modern Regulatory Approvals?
Regulatory authorities increasingly expect manufacturers to provide structured summaries of extractables and leachables data together with process and formulation information to facilitate efficient review and reduce the likelihood of deficiency letters. Structured data management supports reliable extractable-to-leachable correlation, demonstrating that each detected leachable can be traced to an upstream material source or a characterized degradation pathway.
Regulatory review data indicate that E&L-related deficiencies contribute to delays or rejections in approximately twenty percent of drug applications. Common deficiencies include inadequate extraction study design, inaccurate AET calculations, and incomplete evaluation of packaging and manufacturing systems.
To address these concerns, the U.S. FDA Office of Pharmaceutical Quality (OPQ) and Office of Process and Facilities (OPMA) recommend submission of structured E&L datasets within eCTD Module 3.2.R or 3.2.P using standardized spreadsheet (.xlsx) formats.
[ Standardized Technical Dossier (eCTD 3.2.R) ]
│
┌──────────────────────┼──────────────────────┐
▼ ▼ ▼
[ Material Profiling ] [ Method Validation ] [ Toxicological PDE ]
- Polymer Chemistry - LOQ below AET - TTC Comparisons
- Supplier Packages - Recovery Studies - In-Silico QSARThis structured presentation allows regulators to assess E&L findings in the context of formulation attributes, manufacturing variables, and toxicological thresholds. A key aspect of this evaluation is confirmation of extractable-to-leachable correlation.
Any organic leachable detected above the AET at the end of a product’s shelf life should be traceable to a parent extractable or a known degradation product previously characterized during controlled extraction studies.
Furthermore, risk assessments should include evaluation of secondary packaging contamination sources. Volatile organic compounds, including adhesive constituents, plasticizers, and ink photoinitiators such as isopropylthioxanthone, may permeate semi-permeable plastic containers, including LDPE infusion bags, and subsequently contaminate the drug product.
By compiling a comprehensive and structured E&L dossier encompassing primary packaging, manufacturing systems, and secondary packaging components, biopharmaceutical manufacturers can demonstrate a robust control strategy and reduce the likelihood of receiving costly Complete Response Letters (CRLs).
Conclusion
A comprehensive extractables and leachables program serves as a foundational component of biopharmaceutical quality control, protecting product integrity, patient safety, and regulatory timelines. Effective management of Extractables and Leachables in biologic formulations depends on advanced mass spectrometry technologies combined with rigorous toxicological risk assessment methodologies.
Because biologic drug products are structurally delicate and inherently susceptible to degradation, they represent one of the highest-risk categories for interactions with packaging materials and manufacturing systems. Standardized testing programs must be implemented to identify both organic and inorganic migrants before they affect therapeutic performance, alter critical quality attributes, or provoke unwanted immune responses.
To ensure your development program remains on track, consult the Toxicological Qualification of Leachables.
Successfully navigating this complex regulatory environment and achieving compliance with the requirements of USP 665, USP 1665, and emerging ICH Q3E guidance requires specialized analytical expertise. Organizations seeking to develop scientifically justified AET thresholds, implement tailored E&L testing programs, or qualify single-use bioprocessing systems for regulatory submissions should engage experienced analytical specialists. Biopharmaceutical manufacturers may utilize the ResolveMass Laboratories Inc. Contact Us portal to collaborate with a dedicated scientific team and accelerate development programs with confidence while maintaining regulatory compliance.
Frequently Asked Questions
The primary container closure system remains in continuous contact with the biologic product throughout manufacturing, storage, and distribution. Since biologic formulations often contain aqueous buffers, surfactants, stabilizers, and carefully controlled pH conditions, they can facilitate the migration of chemical substances from packaging materials. Components such as plasticizers, antioxidants, and rubber additives may gradually enter the formulation over time. Even trace levels of these compounds can affect protein stability, product quality, and ultimately patient safety.
Transition metals including iron, copper, chromium, cobalt, and nickel can leach from manufacturing equipment, stainless steel surfaces, or container materials. Once present in a biologic formulation, these metals participate in redox reactions that generate reactive oxygen species (ROS), including hydroxyl radicals. These highly reactive species can oxidize vulnerable amino acid residues, causing structural changes, aggregation, and loss of biological activity. Detection and quantification of these metals are typically performed using Inductively Coupled Plasma Mass Spectrometry (ICP-MS), which provides highly sensitive elemental analysis at trace concentration levels.
The initial Analytical Evaluation Threshold (AET) is calculated using toxicological safety limits and patient exposure assumptions, providing a baseline concentration above which compounds must be evaluated. However, analytical instruments do not respond equally to all chemical structures, which can affect concentration estimates during non-targeted screening. To compensate for this variability, the initial AET is modified using an Uncertainty Factor (UF). The resulting adjusted AET provides a more conservative reporting threshold and helps ensure potentially hazardous compounds are not overlooked.
The Uncertainty Factor serves as a correction mechanism that accounts for differences in detector response among unknown compounds. During non-targeted screening, some analytes produce weaker signals than others, which may lead to underestimation of their true concentrations. While a UF of 2 has traditionally been accepted for certain gas chromatography applications, LC-MS analyses often exhibit substantially greater response variability. Studies have shown that higher UF values, sometimes reaching 10, are necessary to reduce the risk of false-negative results and improve detection reliability.
A direct leachables stability study becomes necessary when a risk assessment cannot adequately demonstrate product safety. This typically occurs when extractable compounds exceed established safety thresholds, when toxicological information is incomplete, or when the formulation contains aggressive chemical conditions. Factors such as extreme pH levels, high surfactant concentrations, or the presence of organic solvents can significantly increase the likelihood of chemical migration. Under these circumstances, direct testing is required to confirm the actual levels of leachables present during product storage.
USP General Chapter 665 establishes standardized requirements for extractables characterization of plastic materials used in biopharmaceutical manufacturing systems. USP General Chapter 1665 complements this framework by providing a risk-based methodology for evaluating the suitability of these materials under specific processing conditions. Factors such as contact time, temperature, material composition, and purification capability are assessed during qualification. Together, these chapters help manufacturers demonstrate that single-use components do not adversely affect product quality or patient safety.
No, compliance with pharmacopeial material standards does not eliminate the need for extractables and leachables assessments. Compendial testing primarily evaluates general material characteristics, biological reactivity, and basic safety attributes. However, it does not determine whether specific chemicals may migrate into a drug product under actual manufacturing and storage conditions. Therefore, product-specific E&L studies remain necessary to evaluate potential patient exposure and ensure regulatory compliance.
LC-HRMS platforms such as QTOF and Orbitrap systems provide highly accurate mass measurements that support the identification of unknown compounds present at very low concentrations. These instruments enable precise molecular formula determination and facilitate differentiation between compounds with similar masses. When combined with tandem mass spectrometry and ion mobility techniques, they provide detailed structural information that assists in identifying degradation products, polymer additives, and complex leachable species. This makes LC-HRMS an indispensable tool for comprehensive E&L characterization.
Secondary packaging materials can contribute contaminants even when they do not directly contact the drug product. Volatile and semi-volatile compounds originating from inks, adhesives, coatings, or photoinitiators may migrate through the air space within packaging systems or diffuse through semi-permeable polymer containers. Over extended storage periods, these substances can reach the product and become measurable leachables. For this reason, comprehensive E&L assessments must evaluate the entire packaging configuration rather than only the primary container.
Reference:
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2025, September). ICH Q3E: Guideline for extractables and leachables: Step 2 draft guideline – Released for comments [PowerPoint presentation]. ICH. https://database.ich.org/sites/default/files/ICH_Q3E_Step2_Presentation_2025_0826.pdf
- U.S. Food and Drug Administration. (2024). ANDA submission: Risk-based extractable and leachable quality information [Presentation slides]. U.S. Department of Health and Human Services. https://www.fda.gov/media/183127/download
- European Medicines Agency. (2025, August 18). ICH Q3E extractables and leachables – Scientific guideline. https://www.ema.europa.eu/en/ich-q3e-extractables-leachables-scientific-guideline
- Kaja, R. K. (2025, December 18). Extractables and leachables testing: Driving global standards through dialogue. Quality Matters. U.S. Pharmacopeia. https://qualitymatters.usp.org/extractables-and-leachables-testing-driving-global-standards-through-dialogue

