
Introduction
Obtain regulatory approval for IND, NDA, and BLA submissions with the support of professional Extractables & Leachables (E&L) Consulting Services that establish a scientifically robust material safety and compatibility profile. These specialized consulting services integrate comprehensive chemical screening, toxicological risk assessment, and compendial compliance to detect and evaluate potential contaminants that may migrate from primary packaging, secondary packaging, or bioprocess systems into drug formulations. For pharmaceutical and biopharmaceutical organizations throughout North America, partnering with an experienced Contract Research Organization (CRO) such as ResolveMass Laboratories Inc., a USFDA-registered (FEI: 3042696771), Health Canada-licensed (DEL: 3-002945-A), and ISO 9001:2015 certified laboratory, enables the generation of high-quality, audit-ready data packages that facilitate efficient regulatory review. As the global E&L testing market is expected to expand from 1.13 billion in 2024 to approximately 3.57 billion by 2033, sponsors must navigate an evolving regulatory landscape that increasingly emphasizes Quality Risk Management (QRM) in accordance with ICH Q9 principles. Conducting proactive material assessments significantly reduces the likelihood of late-stage clinical failures, formulation instability, and regulatory compliance issues.
To avoid costly setbacks in your regulatory journey, uncover the common root causes of failed extractables and leachables (E&L) studies and learn how to proactively prevent them.
Share via:
Article Summary:
- Extractables & Leachables (E&L) studies are essential for IND, NDA, and BLA submissions because they demonstrate that packaging materials and manufacturing components do not introduce harmful contaminants into pharmaceutical or biologic products.
- Regulatory expectations become more rigorous as products advance through development. Early-stage IND submissions typically require risk assessments and extractables data, while NDA and BLA applications demand validated, long-term leachables studies supported by comprehensive stability data.
- A structured E&L program follows a stepwise workflow beginning with material selection and extractables testing, followed by Analytical Evaluation Threshold (AET) determination, targeted leachables studies, and toxicological risk assessment to ensure patient safety.
- Well-designed stability studies are critical and should evaluate multiple storage conditions, sampling timepoints, container orientations, and worst-case fill volumes to accurately monitor potential chemical migration throughout the product’s shelf life.
- Reliable chemical characterization depends on multiple analytical technologies, including GC-MS, HS-GC-MS, UPLC-HRMS, and ICP-MS, to identify volatile, non-volatile, and inorganic contaminants at extremely low concentrations.
- Any detected compounds exceeding the Analytical Evaluation Threshold (AET) must undergo a scientific toxicological evaluation using established regulatory principles, exposure limits, and risk assessment methodologies to determine their safety for patients.
- A submission-ready E&L dossier combines scientific, analytical, and toxicological evidence into a comprehensive regulatory package that supports product quality, regulatory compliance, and successful approval while minimizing risks during development and commercialization.

Regulatory Scrutiny in IND, NDA, and BLA Filings
Sponsors are required to include comprehensive extractables and leachables information within the Container Closure System (CCS) section of IND, NDA, and BLA submissions to meet global regulatory expectations. Regulatory agencies, including the USFDA and EMA, carefully assess these submissions to verify that packaging components and manufacturing materials satisfy stringent chemical safety requirements. According to 21 CFR 211.94(a) and 21 CFR 600.11(h), drug containers and closure systems must not be reactive, additive, or absorptive in a manner that could adversely affect the safety, identity, strength, quality, or purity of the drug product. Likewise, EudraLex Volume 4, Part 1, Chapter 3 requires that production equipment contacting drug products must not introduce chemical hazards during manufacturing or storage.
Biopharmaceutical products submitted through BLAs are particularly susceptible to the effects of migrating contaminants. Leachables may trigger protein conformational changes, promote protein aggregation, or chelate inorganic species, thereby directly affecting product stability and patient safety. As a result, regulatory expectations increase according to the route of administration, duration of therapy, and intended patient population. While an initial risk assessment supported by extractables data may be adequate for early clinical development under an IND submission, NDA and BLA approvals require comprehensive, validated leachables studies performed throughout the entire stability program.
Ensure continuous product safety and compliance across your product’s lifecycle by implementing robust leachables monitoring during stability studies for your regulatory submissions.
The regulatory expectations differ considerably depending on the stage of product development:
| Submission Type | Regulatory Stage | Principal E&L Testing Requirement |
|---|---|---|
| IND (Investigational New Drug) | Early-stage clinical development | Material risk assessment, supplier characterization data, and preliminary controlled extractables profiles. |
| NDA (New Drug Application) | Marketing authorization for small-molecule products | Validated, stability-indicating leachables data generated from a minimum of three commercial-scale batches. |
| BLA (Biologics License Application) | Marketing authorization for biologic products | Comprehensive chemical characterization of both container closure systems and bioprocess components, supported by biological safety evaluations. |
Compendial Standards and Testing Workflows
The planning and execution of extractables and leachables studies should follow recognized compendial standards, including USP , USP , and the developing ICH Q3E guideline. These frameworks provide a systematic pathway that progresses from worst-case laboratory extraction studies to realistic assessments of patient exposure under actual product-use conditions. USP establishes the scientific principles for generating a material-specific extractables profile using aggressive laboratory extraction conditions. In contrast, USP describes the appropriate design, execution, and interpretation of leachables studies conducted under simulated or actual storage conditions.
The emerging ICH Q3E guideline further aligns international regulatory expectations by harmonizing the evaluation of both organic and inorganic impurities. It establishes an integrated approach that incorporates risk assessment, risk control, and continuous lifecycle risk management throughout product development and commercialization.

To meet regulatory expectations, extraction study designs should be specifically tailored to the physicochemical characteristics of the drug formulation. For example, aqueous extraction studies for injectable drug products commonly utilize purified water, physiological saline, and acidic or basic extraction media to simulate extreme formulation pH conditions. These studies are generally conducted at temperatures ranging from 40°C to 60°C for a minimum period of 14 to 28 days to maximize the identification of potential extractable compounds.
Optimize your laboratory extraction design and maximize impurity detection when you discover how to select the correct solvents for extractables studies based on your formulation.
Designing Stability Programs with Extractables & Leachables (E&L) Consulting Services
The incorporation of professional Extractables & Leachables (E&L) Consulting Services is essential for establishing a compliant, stability-indicating leachables monitoring program capable of tracking compound migration throughout the product’s intended shelf life. Continuous stability monitoring enables sponsors to identify critical migration trends at an early stage before they develop into significant quality or regulatory concerns. Standard stability studies are performed under multiple storage conditions and container configurations to ensure a complete evaluation of product safety.
Timepoint Pull Schedule: Leachables should be evaluated at predefined intervals, including baseline (T = 0), 6 months (accelerated endpoint for IND assessments), 12 months, 18 months, and 24 months or beyond for long-term shelf-life confirmation. High-risk parenteral products frequently require additional early evaluations at the 1-month and 3-month timepoints.
Sample Orientation: Stability protocols should include both upright and inverted (or horizontal) storage orientations to maintain continuous direct contact between the drug product and elastomeric stoppers or container closure seals throughout the study period.
Fill Volume Ratios: Testing should be performed using the smallest fill-volume configuration because it represents the worst-case condition, resulting in the highest surface-area-to-volume ratio and increasing the potential concentration of leachable compounds.
Accelerate your time to market and ensure the safety of critical injectables by partnering with our experts for specialized E&L testing for pre-filled syringes.
To preserve analytical accuracy during stability studies, laboratories should conduct matrix-spike recovery experiments using the actual drug product at baseline (T = 0). This step is particularly important because formulations containing surfactants, antioxidants, or preservatives may interfere with the detection of low-level migrating compounds. Furthermore, the analytical Limit of Quantitation (LOQ) should remain well below the calculated AET rather than simply meeting the generic Qualification Threshold (QT), ensuring reliable detection and accurate quantification of all relevant leachables throughout the stability program.
Guarantee the safety and functionality of your combination products when you leverage our tailored extractables and leachables testing for autoinjectors.
AET Derivation and Analytical Response Factor Uncertainty
The Analytical Evaluation Threshold (AET) is a scientifically derived reporting threshold that establishes the concentration at which an extractable or leachable compound must be identified and subjected to toxicological qualification. Developing a scientifically justified AET helps prevent the under-reporting of potentially hazardous impurities while ensuring that analytical efforts remain focused on compounds capable of posing meaningful risks to patients.
The calculation process begins by selecting the appropriate Safety Concern Threshold (SCT), which represents the daily exposure level below which a migrating compound is not expected to present systemic toxicity or mutagenic risk. For orally inhaled and nasal drug products (OINDP), the Product Quality Research Institute (PQRI) recommends an SCT of 0.15 µg/day. For parenteral and ophthalmic drug products (PODP), the established SCT is 1.5 µg/day.
Navigate the complexities of analytical reporting thresholds by mastering the calculations for the AET in extractables and leachables studies.
The mathematical expression used to calculate the Estimated AET is shown below using LaTeX notation:
Estimated AET (μg/mL) = SCT (μg/day) Maximum Daily Dose (mL/day) × Doses per Container Total Volume in Container (mL)
Following the calculation of the Estimated AET, sponsors should determine the Final AET by applying an analytical Uncertainty Factor (UF) to compensate for variability in chromatographic Relative Response Factors (RRFs) between target analytes and the internal reference standard.
Final AET = Estimated AET × UF
Alternatively, the Final AET may be determined by incorporating a defined analytical uncertainty margin directly into the calculation:
Final AET = Estimated AET − (Analytical Uncertainty)
The UF is established using relative response factor databases. A default UF of 0.5, representing a 50% analytical uncertainty margin, is widely accepted for many applications. However, highly variable analytical matrices may require a statistically justified uncertainty factor derived from experimental response factor databases to minimize the likelihood of false-negative results.
Address the strict regulatory demands of specialized delivery systems by exploring our comprehensive E&L testing framework for inhalation and nasal drug products.
Orthogonal Analytical Platforms and Instrument Parameters
Comprehensive chemical characterization requires the use of multiple orthogonal chromatographic and mass spectrometry techniques to ensure the detection of volatile, semi-volatile, non-volatile, and inorganic compounds. Employing highly sensitive, calibrated analytical instrumentation enables laboratories to detect potential contaminants at ultra-trace concentrations that remain below the calculated AET.
Advanced analytical laboratories employ specialized chromatographic configurations to generate accurate and reproducible datasets across diverse chemical classes.
Volatile Organic Compounds (VOCs): VOCs are analyzed using HS-GC-MS. A typical compendial headspace configuration incorporates a Shimadzu TQ8050NX triple quadrupole mass spectrometer coupled with an AOC-20S auto-injector and a DB-624 column measuring 60 m × 0.25 mm × 1.4 µm. Standard operating conditions include vial equilibration temperatures between 75°C and 85°C for approximately 20 minutes, Helium carrier gas flowing at 1.5 mL/min, and Electron Impact (EI) ionization operating in full-scan acquisition mode across a mass range of 35–300 m/z.
Semi-Volatile Organic Compounds (SVOCs): SVOCs are evaluated using direct-injection GC-MS or GC-FID methods to identify and quantify plasticizers, antioxidants, slip agents, and other semi-volatile additives commonly associated with packaging materials.
Non-Volatile Organic Compounds (NVOCs): NVOCs are characterized using UPLC-HRMS platforms, including QTOF or Orbitrap systems equipped with soft Electrospray Ionization (ESI), allowing accurate identification of polar compounds, polymer additives, and high-molecular-weight oligomers.
Inorganic and Elemental Species: Inorganic contaminants are screened using ICP-MS in accordance with ICH Q3D guidelines to detect metallic catalysts, glass-derived elemental species, and heavy metals at sub-parts-per-trillion concentrations.
| Analytical Method | Target Compound Class | Destructive? | Focus Keyword Alignment |
|---|---|---|---|
| HS-GC-MS | Volatile Organics and Gases | Yes | Used in specialized E&L services. |
| GC-MS / FID | Semi-Volatile Organic Compounds | Yes | Quantitative profiling using reference materials. |
| UPLC-HRMS | Polar and Non-Volatile Organic Compounds | Yes | Critical for complex biologic packaging evaluations. |
| ICP-MS | Trace Metals and Inorganic Species | Yes | Standard elemental impurity screening following ICH Q3D. |
Make informed decisions for your material screening strategy by understanding the critical differences between GC-MS vs LC-MS in extractables and leachables testing.
Toxicological Qualification and Structure-Based Risk Assessments
Chemical migrants detected at concentrations exceeding the calculated AET must undergo a structured toxicological evaluation to establish whether they are safe at the intended clinical dose and route of administration. For every regulatory submission, these toxicological assessments should be documented within Module 2.6.6.8 (Toxicology Written Summary) of the regulatory dossier. Whenever a leachable exceeds the AET, the toxicological evaluation proceeds through a systematic hierarchy of risk assessment.
Mutagenic Alerts (ICH M7): Any leachable containing a structural alert associated with mutagenicity should be restricted to a maximum chronic daily exposure of 1.5 µg/day. For acute or short-term therapeutic use, exposures up to 120 µg/day may be considered acceptable when scientifically justified.
Non-Genotoxic Impurities: Non-mutagenic leachables present at exposure levels exceeding 5 µg/day require a comprehensive toxicological risk assessment to establish appropriate safety margins and determine acceptable patient exposure.
ICH Q3E Hazard Categorization: The draft ICH Q3E guideline categorizes leachables into three hazard classes based on toxicological potency.
Class 1 (High Concern): This category includes known mutagens and highly potent non-mutagenic compounds with acceptable daily intake limits below 1.5 µg/day, such as Benzo(a)pyrene and Bisphenol A. These substances should either be avoided entirely or maintained below highly restrictive exposure limits.
Class 2 (Default): This category includes compounds for which standard systemic toxicity thresholds, including the Threshold of Toxicological Concern (TTC) and Qualification Threshold (QT), are considered sufficiently protective for patient safety.
Class 3 (Low Potency): This category consists of compounds demonstrating relatively low toxicological concern and possessing chronic parenteral Permitted Daily Exposure (PDE) values of 1 mg/day or greater. Representative examples include BHT, erucamide, 4-tert-amylphenol, C8-C22 fatty acids, and the rubber oligomer C21H40. These substances may be qualified at comparatively higher exposure limits when supported by appropriate toxicological evidence.
When compound-specific toxicological information is unavailable or limited, toxicologists establish acceptable exposure limits by calculating a Permitted Daily Exposure (PDE) using the Point of Departure (POD) obtained from animal toxicology studies and applying appropriate safety uncertainty factors. In situations where published toxicological data are unavailable, in silico QSAR modeling and toxicological read-across assessments using structurally related analogs are employed to support scientifically justified safety evaluations.
Safeguard patient health and support your submission dossier when you utilize our advanced processes for the toxicological qualification of leachables.
Compliance Under USP and for Single-Use Systems
The implementation of USP , effective May 1, 2026, requires biopharmaceutical manufacturers to characterize and qualify polymeric manufacturing components that come into direct contact with drug substances or process intermediates. This mandatory compendial chapter addresses an important product safety concern by requiring the identification and assessment of process equipment-related leachables (PERLs) that may migrate from filters, tubing, connectors, bioreactors, mixing bags, or other single-use components into the product stream.
Compliance is achieved through the risk-based evaluation framework described in the companion chapter, USP . Manufacturers are expected to perform a comprehensive, multidimensional risk assessment that evaluates process temperatures, duration of material contact, formulation pH, solvent polarity, and the effectiveness of downstream purification operations, including chromatography columns and filtration systems, that may reduce or eliminate potential contaminants before the final drug product is obtained.
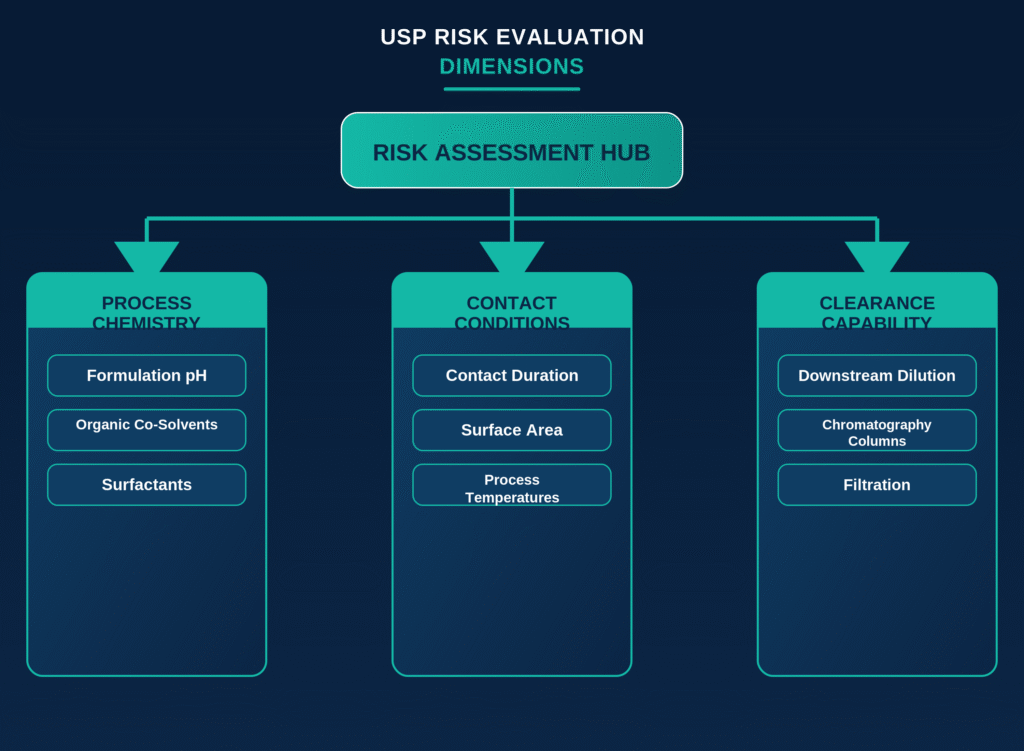
Polymeric components identified as presenting a higher level of risk should undergo controlled extraction studies using appropriate model solvents. These studies should then be followed by advanced chemical characterization to verify that no process equipment-related leachables migrate into the product stream at concentrations that could present toxicological concerns.
Mitigate chemical migration risks right from the start by identifying and selecting the optimal low leachables packaging materials for your critical therapies.
Strategic Execution of a Defensible Regulatory Dossier
A regulatory submission-ready Extractables and Leachables (E&L) dossier should integrate materials science, worst-case extractables data, validated stability-indicating leachables results, and scientifically justified toxicological evaluations into a comprehensive and well-supported regulatory narrative. Engaging experienced Extractables & Leachables (E&L) Consulting Services helps ensure that all analytical findings and supporting documentation are compiled into a scientifically defensible, audit-ready submission package suitable for regulatory review.
A comprehensive E&L regulatory submission package should include the following five essential components:
Container Closure Description: A thorough description of the physical characteristics, chemical composition, and construction of all primary and secondary packaging components that come into contact with the drug product.
Scientific Extraction Justification: A detailed scientific rationale supporting the selection of extraction solvents, extraction temperatures, and extraction durations used to establish the worst-case extractables profile.
Comprehensive Leachables Data: Complete stability datasets documenting the identity of each detected migrant, corresponding sampling timepoints, measured concentrations throughout the stability program, calculated daily patient exposures, and direct comparisons with the calculated Analytical Evaluation Threshold (AET).
Extractables-to-Leachables Correlation: A scientifically supported discussion demonstrating the relationship between observed leachables and their originating material sources, while confirming that no unidentified or non-source-correlated chromatographic peaks remain unexplained.
Toxicological Risk Assessments: Signed toxicological safety evaluations prepared using established toxicity limits, Thresholds of Toxicological Concern (TTCs), or calculated Permitted Daily Exposures (PDEs) for every leachable detected above the established study AET.
Conclusion
Successfully obtaining regulatory clearance for IND, NDA, and BLA submissions depends on utilizing experienced Extractables & Leachables (E&L) Consulting Services to develop a scientifically rigorous, toxicologically sound, and fully compliant material safety dossier. Sponsors seeking dependable regulatory support benefit from collaborating with a specialized, USFDA-registered and Health Canada-licensed Contract Research Organization (CRO) capable of generating the high-quality mass spectrometry and chemical characterization data required to satisfy regulatory expectations and inspection requirements. Implementing a proactive, lifecycle-based strategy for material characterization minimizes the likelihood of late-stage clinical failures while supporting the continued safety, stability, compatibility, and quality of pharmaceutical and biopharmaceutical products throughout their lifecycle.
To discuss your product, material selection, and customized testing requirements with our PhD-level mass spectrometry and regulatory compliance specialists, please visit our Contact Us page.
Frequently Asked Questions
The final Analytical Evaluation Threshold (AET) is determined by first calculating an estimated threshold using the applicable Safety Concern Threshold (SCT) and the maximum daily product dose. An analytical Uncertainty Factor (UF) is then applied to account for differences in detector response and analytical variability. This scientifically justified threshold ensures that compounds capable of impacting patient safety are consistently detected, evaluated, and reported during analytical testing.
The AET and the Limit of Detection (LOD) serve different purposes within an E&L study. The AET is a toxicology-based reporting threshold derived from patient exposure considerations, whereas the LOD represents the lowest concentration that an analytical instrument can reliably detect. A compound may be measurable below the AET, but unless it exceeds the reporting threshold, it generally does not require full identification or toxicological qualification.
USP and USP require manufacturers to evaluate polymeric single-use components that come into contact with drug substances during manufacturing. Components such as bioprocessing bags, tubing, filters, and connectors must undergo extraction studies and risk assessments to determine whether process equipment-related leachables (PERLs) could migrate into the product. Compliance with these standards helps ensure product quality, process reliability, and patient safety throughout manufacturing.
Prefilled syringes frequently contain UV-cured acrylate adhesives within needle assemblies and related components. If the curing process is incomplete, residual monomers such as isobornyl acrylate (IBOA) may migrate into the drug formulation over time. These compounds have the potential to affect protein stability, promote aggregation, or trigger immune-related responses, making their identification and control an important aspect of product safety.
Metallic leachables are routinely monitored using Inductively Coupled Plasma Mass Spectrometry (ICP-MS), which provides highly sensitive detection of elemental impurities. Stability studies evaluate whether metals originating from packaging components, manufacturing equipment, or catalysts migrate into the drug product throughout its shelf life. The measured concentrations are then compared with the Permitted Daily Exposure (PDE) limits established under ICH Q3D to confirm patient safety.
An extractables-to-leachables correlation is generally expected when leachables are detected in the finished pharmaceutical product, particularly in NDA and BLA submissions. This assessment links each detected leachable to its originating material or packaging component and demonstrates that the compound has been properly identified and evaluated. Providing this correlation strengthens the scientific justification of the E&L program and supports regulatory confidence in the submitted data.
Biologic therapies contain large, structurally complex molecules that are highly sensitive to their surrounding chemical environment. Even trace levels of organic or inorganic leachables may alter protein folding, increase aggregation, or reduce molecular stability. Such changes can affect product efficacy and may elevate the risk of unwanted immunogenic responses, making comprehensive E&L studies particularly important for biologic products.
A standard Extractables & Leachables program generally requires approximately 8 to 12 weeks to complete the extractables phase, including study design, sample preparation, analytical method development, and chemical characterization. When targeted leachables studies are required, they are usually conducted alongside long-term GMP stability studies. These evaluations continue over multiple timepoints, including 6, 12, 18, and 24 months or longer, depending on the intended product shelf life.
When compound-specific toxicological information is limited or unavailable, toxicologists rely on multiple scientific approaches to evaluate safety. The assessment typically begins with screening for mutagenic structural alerts in accordance with ICH M7 guidance. Additional evaluations may include in silico QSAR modeling, read-across analyses using structurally related compounds, and calculation of a scientifically justified Permitted Daily Exposure (PDE). These methods provide a reliable framework for establishing acceptable exposure limits in the absence of published toxicological data.
Reference:
- European Medicines Agency. (2025). ICH Q3E extractables and leachables – Scientific guideline. https://www.ema.europa.eu/en/ich-q3e-extractables-leachables-scientific-guideline
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2025, September). ICH Q3E: Guideline for extractables and leachables—Step 2 draft guideline: Presentation [PowerPoint presentation]. https://database.ich.org/sites/default/files/ICH_Q3E_Step2_Presentation_2025_0826.pdf
- Di Bello, A., Caporali, A., & colleagues. (2025). Extractables and leachables in pharmaceutical products: Current regulatory perspectives and analytical strategies. Pharmaceutics. Advance online publication. https://pmc.ncbi.nlm.nih.gov/articles/PMC12283826/
- United States Pharmacopeia. (2024, August 26). USP updates on extractables and leachables (E&L) [Conference presentation]. https://www.usp.org/sites/default/files/usp/document/events-training/08%20-%20E%26L%20Updates%20from%20USP.pdf

