
Introduction
Establishing a validated GMP Extractables and Leachables Testing program is an essential aspect of modern pharmaceutical development. It ensures that chemical compounds capable of migrating from manufacturing equipment or container-closure systems do not compromise patient safety, product efficacy, or long-term drug stability. As regulatory expectations continue to evolve through initiatives such as the draft ICH Q3E guideline and updated pharmacopeial chapters, pharmaceutical organizations are expected to implement scientifically justified risk assessments supported by robust analytical methodologies. ResolveMass Laboratories Inc., an ISO 9001:2015 certified Contract Research Organization (CRO) and Contract Development and Manufacturing Organization (CDMO) holding a Health Canada Drug Establishment Licence (DEL 3-002945-A) and USFDA registration (FEI 3042696771), provides advanced analytical testing and custom synthesis services under full GMP compliance for pharmaceutical and biotechnology innovators. By utilizing high-resolution mass spectrometry, nuclear magnetic resonance spectroscopy, and specialized polymer characterization technologies, organizations can generate comprehensive analytical data packages that satisfy demanding global regulatory requirements while accelerating product development timelines and market entry.
Ensure compliance and safety by reviewing our rigorous approach to data integrity in extractables and leachables testing.
Share via:
Article Summary:
- GMP Extractables and Leachables (E&L) Testing is essential for identifying chemicals that may migrate from packaging materials or manufacturing components into drug products, helping protect patient safety, product quality, and regulatory compliance throughout the product lifecycle.
- Global regulatory expectations are guided by USP chapters such as USP <661.1>, <661.2>, <665>, <1663>, <1664>, and <382>, together with the draft ICH Q3E guideline, which promote science-based risk assessments, lifecycle management, and comprehensive material qualification.
- USP <665> and USP <1665> introduce a structured risk-based qualification process for polymeric manufacturing components. Manufacturers must evaluate process-contact materials based on factors such as fluid chemistry, temperature, exposure duration, and downstream purification before determining the appropriate extractables testing strategy.
- The Analytical Evaluation Threshold (AET) provides a scientifically justified reporting limit for extractables and leachables. It is calculated using the Safety Concern Threshold (SCT), product-specific dosing information, and an analytical uncertainty factor to ensure compounds with potential toxicological significance are reliably detected.
- USP <382> expands regulatory expectations by requiring functional testing of complete elastomeric container-closure systems, including fragmentation, penetration force, self-sealing performance, break-loose and glide force, and container closure integrity, ensuring reliable performance under real-use conditions.
- Comprehensive chemical characterization relies on orthogonal analytical techniques, including GC-MS, LC-MS/HRMS, NMR, and ICP-MS, enabling accurate identification of volatile, non-volatile, polymer-related, and elemental impurities that may migrate into pharmaceutical products.
- A successful GMP E&L program combines material risk assessment, controlled extraction studies, validated analytical methods, real-time leachables monitoring, toxicological qualification, and lifecycle risk management to generate inspection-ready data that supports global regulatory submissions and long-term product quality.
Regulatory Standards and Subchapters Governing Packaging and Manufacturing Systems
Regulatory oversight for pharmaceutical container-closure systems and manufacturing components is established through a harmonized framework of United States Pharmacopeia (USP) chapters and International Council for Harmonisation (ICH) quality guidelines. Collectively, these regulatory standards ensure that every material with direct or indirect contact with manufacturing intermediates or finished drug products undergoes comprehensive qualification to verify its chemical safety and suitability throughout the product lifecycle.
Compliance Frameworks for GMP Extractables and Leachables Testing
Achieving regulatory compliance requires drug product teams to align their analytical strategies with distinct pharmacopeial pathways based on whether the material is associated with manufacturing processes or packaging systems. This structured regulatory approach enables thorough chemical characterization of materials of construction to identify potential extractables during development, while simultaneously establishing appropriate monitoring strategies for leachables that may migrate into the drug product throughout its shelf life.
To assist pharmaceutical organizations in understanding these regulatory expectations, the relationship between major standards, applicable material categories, and their corresponding compliance objectives is summarized below.
| Standard / Chapter | Material Scope | Core Regulatory Compliance Actions |
|---|---|---|
| USP <661.1> | Plastic Materials of Construction | Establishes identity, biocompatibility, and chemical safety requirements for raw polymeric materials used in pharmaceutical packaging. |
| USP <661.2> | Plastic Packaging Systems for Pharmaceutical Use | Evaluates completed plastic packaging systems, including bottles, caps, blister packs, and other finished container-closure assemblies. |
| USP <665> | Polymeric Manufacturing Components & Systems | Requires standardized extraction studies and risk-based assessments for single-use manufacturing components that contact pharmaceutical process streams. |
| USP <1663> | Extractables Associated with Packaging Systems | Provides scientifically justified, non-prescriptive guidance for designing and conducting controlled extractables studies. |
| USP <1664> Series | Leachables Associated with Packaging Systems | Recommends approaches for designing real-time stability studies to evaluate the migration of chemical compounds into pharmaceutical products throughout their shelf life. |
| ISO 10993-18 | Medical Devices and Combination Products | Specifies chemical characterization requirements and toxicological risk assessments for combination products, including prefilled syringes and related delivery systems. |
| ICH Q3E (Draft) | Global Drug Products and Delivery Devices | Harmonizes international strategies for risk assessment, qualification, and lifecycle control of extractables and leachables. |
The proposed expansion of the USP <1664> series reflects the industry’s transition toward highly targeted evaluations based on the intended route of administration rather than relying on a single generalized testing strategy. Accordingly, pharmaceutical manufacturers are expected to develop stability-indicating leachables methods that address route-specific clinical risks. Examples include pulmonary safety considerations for OINDP (USP <1664.1>), systemic toxicity concerns associated with parenteral products under USP <1664.2>, and localized ocular safety assessments for ophthalmic formulations addressed by USP <1664.3>.
Read our complete guide on specialized regulatory compliance and E&L testing for inhalation and nasal drug products.
Detailed Risk Classification Under USP <665> and USP <1665>
USP <665> and USP <1665> establish a comprehensive, risk-based framework for qualifying polymeric manufacturing components utilized in pharmaceutical and biopharmaceutical manufacturing operations. Officially approved in 2024 and becoming fully enforceable on May 1, 2026, these chapters require manufacturers to demonstrate that plastic process-contact components do not release unacceptable concentrations of process equipment-related leachables (PERLs) into manufacturing process streams.
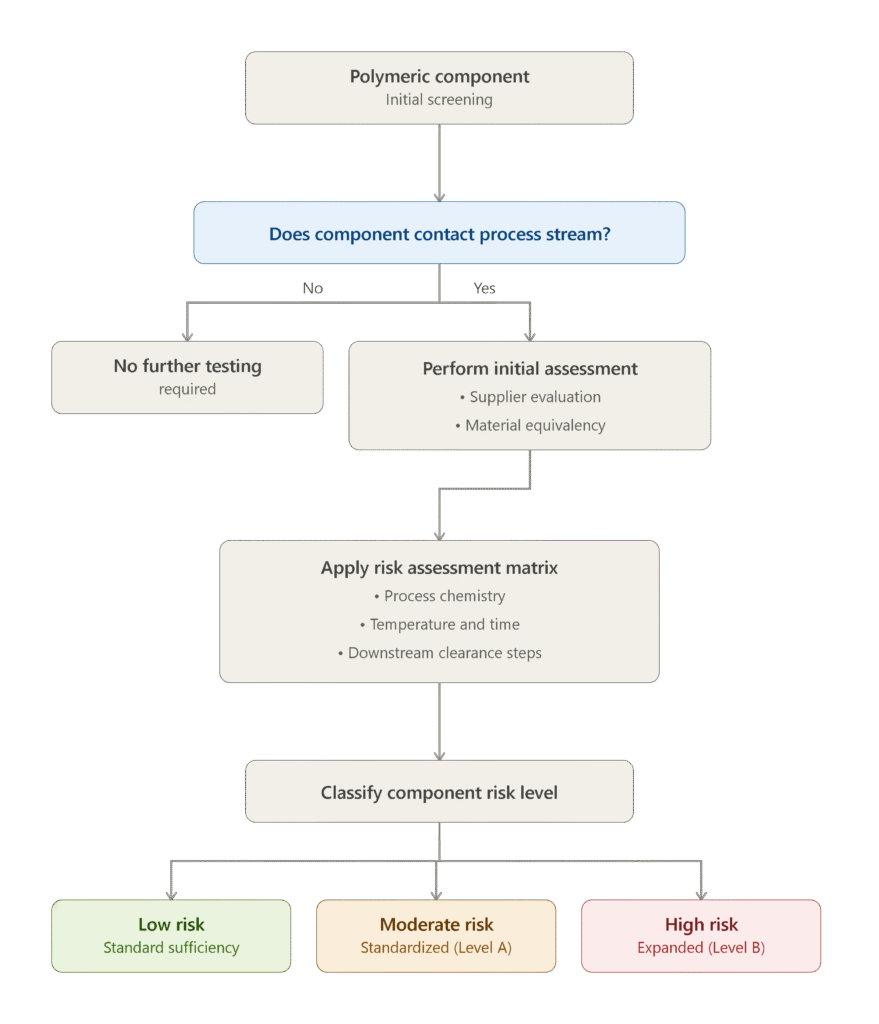
The qualification process begins with an initial evaluation performed in accordance with USP <665> to determine whether the polymeric component comes into direct contact with the pharmaceutical process stream and whether supplier-provided extractables information is sufficient to support material qualification. If existing supplier documentation is incomplete, inadequate, or unavailable, pharmaceutical organizations are required to perform a structured Risk Assessment Matrix to establish the appropriate qualification strategy.
This Risk Assessment Matrix evaluates the potential interaction between the polymeric component and the manufacturing fluid by considering multiple scientific factors, including the following:
Process Fluid Chemistry: This assessment determines the extraction potential of the manufacturing fluid based on its chemical composition. Process solutions containing organic co-solvents, surfactants, or extreme pH conditions generally possess a significantly greater ability to extract additives such as slip agents, plasticizers, antioxidants, and other polymer-associated compounds compared with simple aqueous buffer systems.
Contact Temperature and Duration: This parameter evaluates the thermodynamic and kinetic factors influencing chemical migration. Extended exposure of process fluids to polymeric materials at ambient temperature or processing under elevated thermal conditions substantially increases the likelihood of extractable compound migration compared with short-duration processing or refrigerated filtration operations.
Component Position Relative to Purification Steps: This factor assesses the capability of downstream purification processes to remove or reduce potential contaminants. Polymeric components positioned upstream within manufacturing operations, such as those used before chromatography or ultrafiltration, generally present a lower overall product risk because subsequent purification stages effectively eliminate or dilute many PERLs. In contrast, components used after purification or during final fill-finish operations represent substantially higher risk because any released leachables have a direct pathway into the finished pharmaceutical dosage form.
The cumulative score generated through this Risk Assessment Matrix classifies each polymeric component as presenting low, moderate, or high risk. Components categorized as low risk may be qualified through existing supplier documentation, material certifications, or previously established chemical safety information. Components classified as moderate or high risk require formal laboratory evaluation using the standardized extraction procedures described in USP <665>. These extraction protocols require testing with three standardized model solvents—an acidic solution (pH 3), an alkaline solution (pH 10), and an organic-containing solvent (for example, 50% ethanol)—under prescribed surface-area-to-volume ratios and elevated extraction temperatures. This scientifically standardized approach generates a representative extractables profile that supports comprehensive chemical characterization and regulatory compliance.
Learn how to select the ideal scientific conditions and solvents for extractables studies.
Mathematical Derivation of the Analytical Evaluation Threshold
The Analytical Evaluation Threshold (AET) represents the minimum concentration at which an extractable or leachable compound must be detected, identified, and subjected to toxicological qualification. Incorporating the AET into analytical studies replaces the traditional “detect as low as possible” philosophy with a scientifically justified, risk-based reporting threshold. This approach ensures that analytical investigations remain aligned with patient safety while maintaining consistency with current regulatory expectations.
The Mathematical Formulation of AET and Uncertainty Factors
The final AET is mathematically derived by converting a dose-based Safety Concern Threshold (SCT) into a product-specific concentration limit and then adjusting that value to account for analytical variability through an Uncertainty Factor (UF). This calculation establishes a direct connection between clinical exposure limits and analytical method capability, ensuring that any extractable or leachable capable of exceeding the established safety threshold can be reliably detected during analytical screening.
The derivation is performed through a structured, stepwise calculation process.
Step 1: Determination of the Estimated AET (AETest)
The initial step converts the dose-based safety threshold into a concentration limit specific to the pharmaceutical product. For liquid drug products packaged within container-closure systems, the calculation is expressed as:
AETest = (SCT × Nlabeled) / (Ddaily × Vtotal)
Where:
- SCT is the Safety Concern Threshold, representing the maximum acceptable daily intake below which a leachable is considered to present negligible carcinogenic or non-carcinogenic risk. For parenteral drug products, the SCT is generally 1.5 µg/day, while orally inhaled and nasal drug products (OINDP) require the more conservative threshold of 0.15 µg/day.
- Nlabeled represents the total number of labeled doses contained within the packaging or delivery system.
- Ddaily is the maximum number of doses administered to the patient each day.
- Vtotal is the total volume or mass of the pharmaceutical formulation contained within the package.
For products delivered continuously or in fluid-dose configurations, the equation may be simplified by directly relating the Safety Concern Threshold to the Maximum Daily Dose (MDD):
AETest = SCT / MDD
where MDD is expressed in mL/day or g/day, producing an estimated threshold in µg/mL or µg/g, respectively.
Step 2: Calibration of the Analytical Uncertainty Factor (UF)
Chromatographic screening techniques, including GC-MS and LC-MS, generally rely on a limited number of internal reference standards while simultaneously screening for numerous unknown extractable and leachable compounds. Because individual analytes exhibit different Relative Response Factors (RRFs), analytical response variability can result in underestimation of certain compounds. To compensate for this variability, an Uncertainty Factor (UF) is determined or scientifically justified.
A method-specific UF may be established using an experimentally generated Relative Response Factor database according to the following relationship:
UF = 1 / (1 − RSD)
Where:
- RSD is the Relative Standard Deviation of the response factors obtained from a representative library of target compounds analyzed under identical chromatographic and instrumental conditions.
Alternatively, when response factors are assumed to follow a Gaussian distribution, the Uncertainty Factor may be determined using the following statistical relationship:
UF = μ / (1 − (t × σ))
Where:
- μ represents the mean relative response factor.
- σ represents the standard deviation of the relative response factors.
- t represents the Student’s t-distribution factor corresponding to the selected statistical confidence level.
For direct analytical techniques such as GC-MS or GC-FID, where detector response factors are generally consistent across many analytes, a default UF of 2 is widely accepted within the industry. However, LC-MS methods employing electrospray ionization (ESI) exhibit considerably greater variability because ionization efficiency depends strongly on molecular structure and sample matrix effects. Consequently, the justified UF for LC-MS methods is frequently higher, with default values commonly ranging from 4 to 10 unless experimental data support the use of a lower factor.
Step 3: Calculation of the Final AET (AETfinal)
The final reporting threshold applied during chromatographic evaluation is obtained by dividing the estimated threshold by the established Uncertainty Factor:
AETfinal = AETest / UF
The resulting AETfinal becomes the analytical reporting threshold used to evaluate chromatographic data and determine whether individual extractable or leachable compounds require identification and toxicological assessment.
When the calculated UF becomes excessively large (for example, UF > 10), the resulting AETfinal may fall below the analytical method’s Limit of Quantification (LOQ). Under these circumstances, the analytical method may no longer possess adequate sensitivity to reliably detect compounds at the required reporting level. To address this limitation, laboratories must implement scientifically justified solutions such as sample concentration procedures, enhanced extraction techniques, or analytical method optimization to achieve an LOQ that is lower than the calculated final reporting threshold.
Deepen your understanding of this critical safety equation with our guide to AET for extractables and leachables studies.
Elastomeric Component Functional Suitability Under USP <382>
USP <382> establishes comprehensive system-level performance evaluations for elastomeric closures using the fully assembled, final-use container-closure configuration. First introduced in 2020, this general chapter became officially effective on December 1, 2025, with an updated revision taking effect on February 1, 2026. The chapter represents a major regulatory advancement by shifting the focus from testing individual elastomeric materials to evaluating the performance of fully assembled drug delivery systems under conditions representative of actual clinical use.
Historically, elastomeric components—including stoppers, plungers, needle shields, and tip caps—were primarily evaluated for their raw material composition under USP <381>. Under USP <382>, however, pharmaceutical manufacturers are required to demonstrate that these components perform as intended within the completed container-closure system, which is filled either with the actual pharmaceutical formulation or with a scientifically qualified physicochemical surrogate.
The chapter specifies functional performance tests that are tailored to different packaging configurations and intended clinical applications. The primary testing requirements are summarized below.
| Core Functional Test | Relevant Elastomeric Components | Standard Method Parameters | Primary Failure Risks & Quality Impact |
|---|---|---|---|
| Fragmentation | Vial stoppers, infusion bottle seals | Evaluate closure integrity by puncturing the elastomer with a needle or spike under defined access conditions. | Generation of particulate matter that may contaminate the drug product and present significant systemic safety risks to patients. |
| Penetration Force | Multi-dose stoppers, cartridge septa | Measure the maximum force required to completely penetrate the elastomeric closure. | Excessive penetration force may bend needles, reduce usability, and compromise safe product administration. |
| Self-Sealing Capacity | Multi-dose vial closures subjected to repeated access | Puncture the closure followed by validated vacuum decay or pressure decay leak testing. | Loss of sterile barrier integrity, increasing the risk of microbial contamination during repeated use. |
| Break-Loose & Glide Force | Prefilled syringe plungers and cartridge stoppers | Record both the initial activation force and the continuous movement force throughout syringe operation. | Plunger stiction, inconsistent movement, inaccurate dose delivery, and potential failure of auto-injector systems. |
| Inherent Package Integrity | Plungers, tip caps, and elastomeric seals | Perform deterministic Container Closure Integrity Testing (CCIT) in accordance with USP <1207>. | Product leakage, ingress of reactive atmospheric gases, and loss of sterility throughout storage. |
Implementing these functional evaluations introduces additional technical considerations because testing is performed on fully assembled, product-filled systems rather than isolated elastomeric components. Drug formulations containing active pharmaceutical ingredients, surfactants, silicone oil lubricants, or other excipients can significantly influence elastomer friction, sealing behavior, and long-term mechanical performance. These interactions may alter break-loose forces, glide characteristics, and puncture performance over the product’s shelf life.
Furthermore, the February 1, 2026 revision to USP <382> refined the Needle Self-Sealing Capacity procedure by specifying that the maximum number of punctures should correspond to 1.0 times the intended number of clinical access events. This revision aligns the methodology with ISO 11608-3, preventing excessive puncturing that does not accurately represent real-world clinical use. As a result, the updated procedure minimizes artificially induced leakage failures while maintaining scientifically appropriate and regulatory-compliant performance assessments.
Orthogonal Chromatographic and Spectroscopic Screening Methodologies
Comprehensive chemical characterization of complex extractable and leachable mixtures relies on orthogonal analytical strategies that integrate gas chromatography, liquid chromatography, elemental analysis, and advanced mass spectrometry. Because unknown migrant compounds exhibit substantial variation in volatility, polarity, molecular weight, thermal stability, and ionization behavior, no single analytical technique is capable of identifying every potential compound present within an extractables or leachables profile.
To achieve complete characterization during GMP Extractables and Leachables Testing, multiple complementary analytical platforms are employed. The principal methodologies, together with their analytical capabilities and technical applications, are described below.
1. Gas Chromatography-Mass Spectrometry (GC-MS)
GC-MS serves as the primary analytical platform for the separation, detection, and identification of volatile organic compounds (VOC) and semi-volatile organic compounds (SVOC) that may migrate from packaging materials or manufacturing components.
Headspace GC-MS: This technique is widely used for the analysis of volatile compounds, including residual solvents, unreacted monomers, and other low-boiling constituents. Samples are heated under controlled conditions to transfer volatile analytes into the gas phase before chromatographic injection, thereby minimizing matrix interference while improving analytical sensitivity.
Direct Injection GC-MS: This approach is primarily applied to semi-volatile compounds such as plasticizers (phthalates), polymer degradation products, antioxidants, and stabilizers including BHT and Irganox 1010. Standard Electron Ionization (EI) operating at 70 eV generates highly reproducible fragmentation patterns, allowing the acquired spectra to be compared against well-established commercial spectral libraries such as NIST and Wiley for reliable compound identification.
2. Liquid Chromatography-Mass Spectrometry (LC-MS)
LC-MS is the preferred analytical technique for detecting and characterizing non-volatile organic compounds (NVOC), highly polar substances, polymer oligomers, acid scavengers, and slip agents such as erucamide, which are not amenable to gas chromatographic analysis.
Separation and Ionization: High-Performance Liquid Chromatography (HPLC) coupled with ultraviolet/visible (UV-Vis) detection and High-Resolution Mass Spectrometry (HRMS) provides complementary analytical information for complex sample characterization. Ionization is typically achieved using Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI) operated in both positive and negative ionization modes to maximize analyte coverage across diverse chemical classes.
Structural Elucidation: High-resolution mass analyzers, including Time-of-Flight (TOF) and Orbitrap systems, routinely achieve mass accuracies of less than 5 ppm, enabling the determination of empirical molecular formulas for unknown chromatographic peaks. These highly accurate mass measurements can subsequently be compared with specialized databases such as the Eurofins Extractables Index or proprietary in-house spectral libraries to facilitate efficient structural elucidation and compound identification.
Read our technical breakdown comparing the strengths and applications of GC-MS vs LC-MS in extractables and leachables testing.
3. Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
Inductively Coupled Plasma Mass Spectrometry (ICP-MS) is the analytical technique of choice for the quantification of inorganic extractables, toxic elemental impurities, and residual manufacturing catalysts, including lead, antimony, zinc, arsenic, and other trace metals.
ICP-MS utilizes a high-temperature argon plasma to atomize and ionize sample extracts before mass spectrometric analysis, providing exceptionally low detection limits that routinely extend into the parts-per-trillion (ppt) range. This level of analytical sensitivity is essential for performing toxicological evaluations of elemental impurities originating from packaging systems or manufacturing equipment and supports compliance with the permitted daily exposure limits established under ICH Q3D.
Learn how we apply high-sensitivity ICP-MS in extractables and leachables testing for inorganic impurity quantification.
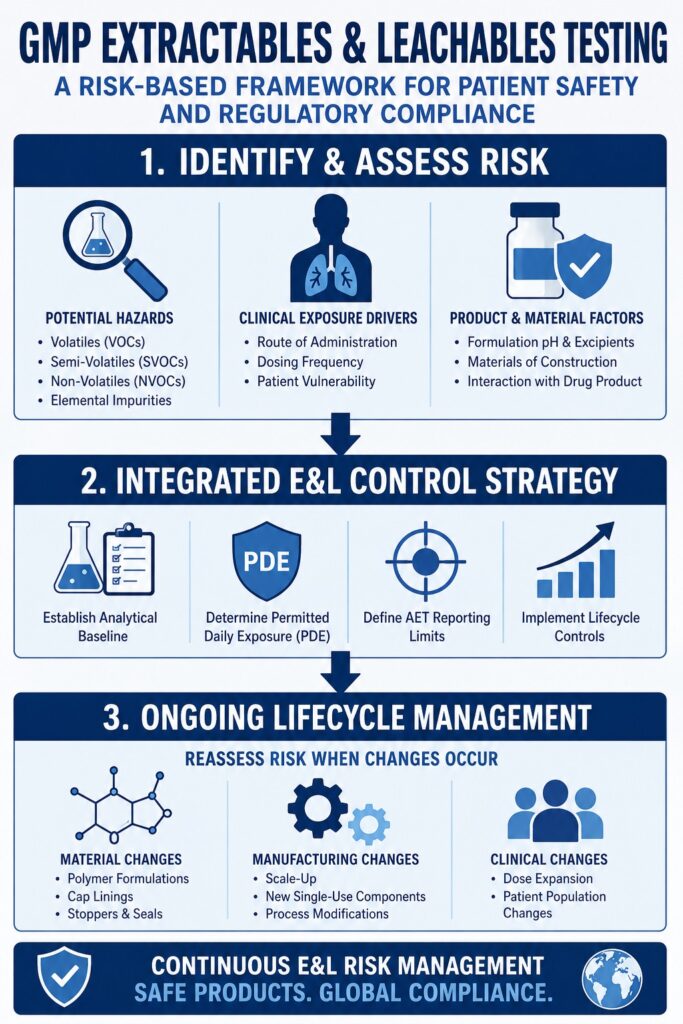
Global Harmonization and Product Lifecycle Risk Control under ICH Q3E
The draft ICH Q3E guideline establishes a globally harmonized quality risk management framework for identifying, evaluating, and controlling extractable and leachable risks throughout the entire pharmaceutical product lifecycle. Officially endorsed as a Step 2 draft in August 2025, the guideline integrates toxicological safety assessments with lifecycle-based quality controls while aligning with the Quality by Design (QbD) principles outlined in ICH Q8, ICH Q9, and ICH Q10.

The ICH Q3E guideline introduces a structured evaluation framework that enables pharmaceutical organizations to systematically classify and control potential leachable risks throughout product development and commercialization.
1. Hazard Identification
The first stage requires drug product teams to compile comprehensive chemical composition information for every product-contact material. This includes evaluating base polymers, additives, processing aids, stabilizers, colorants, antioxidants, and other formulation-related constituents that may contribute to extractable or leachable compounds. Supporting information is gathered through supplier technical dossiers, material certificates, historical extractables databases, and available manufacturing documentation to establish a complete understanding of potential chemical hazards.
2. Risk Analysis and Quantification
Following hazard identification, the likelihood and magnitude of leachable migration are assessed under simulated or actual use conditions. The resulting patient exposure estimates are compared with established toxicological limits, including the Safety Concern Threshold (SCT) and the Qualification Threshold (QT), to determine the appropriate level of risk control.
The guideline broadly categorizes potential compounds into three primary risk classes:
- Class 1 (High Concern): Mutagenic compounds or substances exhibiting significant toxicological concern that must either be eliminated or controlled below compound-specific acceptable exposure limits.
- Class 2 (Moderate Concern): Compounds with established non-mutagenic toxicological effects that require control according to scientifically justified Permitted Daily Exposure (PDE) values.
- Class 3 (Low Concern): Compounds demonstrating substantial toxicological safety margins that can generally be qualified through routine analytical screening supported by appropriate scientific justification.
3. Integrated Control Strategy
Once acceptable exposure limits have been established, pharmaceutical organizations must implement an integrated control strategy to ensure that extractables and leachables remain below hazardous concentrations throughout the entire commercial shelf life of the product. This comprehensive control strategy may incorporate raw material specifications, supplier qualification programs, periodic supplier audits, incoming material quality control testing, and routine monitoring of elastomeric and polymeric components used during manufacturing.
A key principle of ICH Q3E is that Extractables and Leachables (E&L) evaluation is not considered a one-time regulatory submission activity. Instead, it is treated as a continuous lifecycle management process supported by formal change control procedures. Whenever significant post-approval changes occur—such as modifications to elastomer formulations supplied by packaging vendors, manufacturing scale-up activities, implementation of new single-use process components, or expansion of clinical dosing regimens—drug product teams are expected to reassess the existing E&L risk profile and conduct targeted re-evaluation where appropriate. This proactive lifecycle approach helps ensure that the validated state of the pharmaceutical product is maintained throughout commercial manufacturing.
Operationalizing GMP Extractables and Leachables Testing
Successfully implementing a compliant GMP Extractables and Leachables Testing program requires coordinated execution across material selection, analytical method development, laboratory validation, functional performance testing, and toxicological risk assessment. Failure to effectively integrate these activities can increase the likelihood of regulatory observations, compromise product stability, delay product approvals, or introduce unacceptable patient safety risks.
To establish a streamlined and inspection-ready E&L program, pharmaceutical organizations should implement the following structured operational workflow.
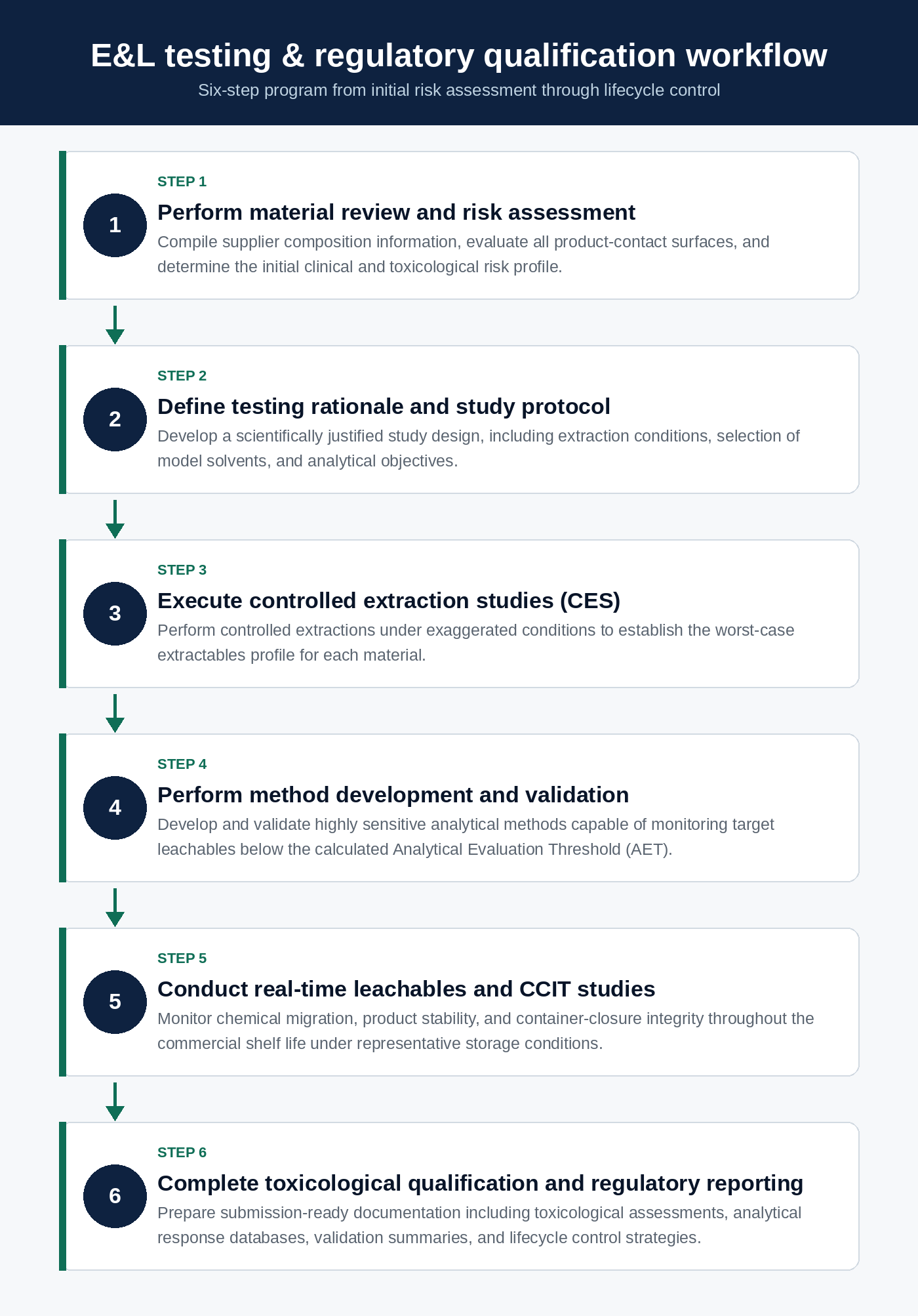
By moving beyond isolated, transaction-based testing activities and adopting this integrated operational framework, pharmaceutical manufacturers can generate robust, scientifically defensible data packages that support global regulatory submissions. Aligning study designs with USP <665>/<1665>, USP <382>, USP <1663>/<1664>, and the draft ICH Q3E guideline helps ensure that regulatory submissions are comprehensive, scientifically justified, and suitable for review by international health authorities.
For customized study designs, analytical method validation, and GMP-compliant testing protocols, drug product teams can contact the ResolveMass team to consult directly with a PhD-level scientist regarding project-specific analytical and regulatory requirements.
Conclusion
Implementing a scientifically robust GMP Extractables and Leachables Testing strategy is fundamental to ensuring the long-term safety, quality, and regulatory compliance of pharmaceutical products. As the industry prepares for the mandatory implementation of USP <665> on May 1, 2026, together with the expanded functional performance requirements introduced under USP <382>, drug product teams must adopt comprehensive, risk-based evaluation approaches rather than relying solely on conventional material testing. Establishing scientifically justified Analytical Evaluation Thresholds (AETs), applying advanced orthogonal analytical technologies for comprehensive chemical characterization, and designing studies that align with the lifecycle risk management principles outlined in the draft ICH Q3E guideline enable organizations to generate reliable, submission-ready data packages. In today’s increasingly stringent regulatory landscape, partnering with experienced analytical laboratories is essential for supporting consistent quality oversight, maintaining lifecycle compliance, and facilitating successful global regulatory submissions.
For customized study designs, analytical method validation, and GMP-compliant testing protocols, drug product teams can contact us here: Contact Page.
Frequently Asked Questions
Compliance with USP <665> and USP <1665> becomes officially enforceable on May 1, 2026. From this date onward, manufacturers using polymeric process-contact materials and single-use systems must comply with the pharmacopeial requirements for risk assessment, extractables characterization, and material qualification. These chapters establish standardized expectations for demonstrating the chemical safety of manufacturing components used in pharmaceutical and biopharmaceutical production.
The Uncertainty Factor (UF) is incorporated into the AET calculation to compensate for differences in analytical response among unknown chemical compounds. Since chromatographic methods often use only a limited number of calibration standards, certain compounds may produce weaker detector responses than others. Applying the UF lowers the reporting threshold, helping ensure that potentially significant extractables or leachables are not overlooked because of reduced analytical sensitivity.
USP <382> shifts the focus from evaluating individual elastomeric materials to assessing the performance of the complete container-closure system in its final-use configuration. Functional testing must be performed using assemblies filled with either the actual pharmaceutical product or a qualified surrogate solution. This approach evaluates characteristics such as fragmentation, self-sealing performance, penetration force, and glide force under conditions that closely resemble actual clinical use.
The PQRI-PODP working group does not recommend a universal Safety Concern Threshold (SCT) for ophthalmic products because these formulations are administered directly to the eye, where localized ocular effects are more relevant than systemic toxicity. Due to limited toxicological data for ocular exposure at concentrations associated with leachables, a single scientifically justified SCT has not been established. As a practical approach, many organizations have historically applied a reporting threshold of 1.0 µg/mL (1 ppm) for ophthalmic leachables.
The February 1, 2026 revision of USP <382> updated the Needle Self-Sealing Capacity test to better align with ISO 11608-3. The revision reduced the required puncture frequency from 1.5 times to 1.0 times the expected number of clinical access events. This modification creates testing conditions that more accurately reflect real-world product use while minimizing artificial leakage failures caused by excessive puncturing during laboratory evaluation.
The Analytical Evaluation Threshold (AET) defines the concentration above which detected compounds require reporting, identification, and toxicological evaluation, whereas the Limit of Quantification (LOQ) represents the lowest concentration that an analytical method can accurately and reliably measure. For an analytical method to be considered suitable, its LOQ should be lower than the calculated AET. If the AET falls below the method’s LOQ, additional method optimization or appropriate sample concentration techniques are required to achieve sufficient analytical sensitivity.
The draft ICH Q3E guideline recommends re-evaluating approved products whenever post-approval changes could influence the extractables or leachables profile or modify patient exposure. Typical triggers include changes to formulation composition, packaging materials, manufacturing equipment, process scale, dosing regimen, duration of therapy, or the intended patient population. Performing a targeted risk assessment after such changes helps ensure that the product continues to meet established safety and quality requirements.
USP <1207> recommends deterministic Container Closure Integrity Testing (CCIT) methods because they provide objective, quantitative, and highly reproducible results. Preferred techniques include vacuum decay, high-voltage leak detection (HVLD), and laser-based headspace gas analysis, all of which deliver reliable measurements of package integrity. Although probabilistic methods such as dye ingress and microbial challenge testing remain available in certain situations, deterministic technologies are generally favored because of their superior sensitivity, repeatability, and scientific robustness.
In most situations, compounds detected below the calculated AET do not require detailed identification or toxicological qualification because the estimated patient exposure remains below established safety thresholds. However, important exceptions exist for compounds of exceptionally high toxicological concern, including nitrosamines, polycyclic aromatic hydrocarbons (PAHs), 2-mercaptobenzothiazole, and other Substances of Very High Concern (SVHC). These compounds require targeted evaluation using highly sensitive and validated analytical methods regardless of whether their concentrations fall below the general AET.
Reference:
- United States Pharmacopeia. (n.d.). Extractables and leachables. U.S. Pharmacopeia. https://www.usp.org/impurities/extractables-and-leachables
- Innovation, Science and Economic Development Canada. (2026, May 8). Canadian life science clinical research and manufacturing capabilities. Government of Canada. https://ised-isde.canada.ca/site/canadian-life-science-industries/en/biopharmaceuticals-and-pharmaceuticals/canadian-life-science-clinical-research-and-manufacturing-capabilities
- European Medicines Agency. (2025, August 18). Draft ICH Q3E guideline for extractables and leachables (EMA/CHMP/ICH/236669/2025). https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-q3e-guideline-extractables-leachables_en.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2025, September). ICH Q3E: Guideline for extractables and leachables—Step 2 draft guideline: Presentation [PowerPoint presentation]. ICH. https://database.ich.org/sites/default/files/ICH_Q3E_Step2_Presentation_2025_0826.pdf
- U.S. Food and Drug Administration. (2025, November). Q3E guideline for extractables and leachables (Draft guidance for industry). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q3e-guideline-extractables-and-leachables
- United States Pharmacopeia. (2017). 〈382〉 Elastomeric closure functionality in injectable pharmaceutical packaging/delivery systems (Pharmacopeial Forum, 43(3)). U.S. Pharmacopeia. https://www.usp.org/sites/default/files/usp/document/workshops/382_elastomeric_closure_functionality_in_injectable_pharmaceutical_packagingdelivery_systems_pf_43_3.pdf

