
Introduction
The development and submission of generic drugs to the U.S. Food and Drug Administration (USFDA) require strict regulatory compliance to ensure product safety, effectiveness, and quality. Impurity Profiling and Characterization for Generic Project plays a crucial role in this process by identifying and evaluating unwanted substances that may originate from raw materials, manufacturing steps, or product degradation. These impurities must be accurately detected, measured, and controlled in accordance with USFDA expectations. Resolvemass Laboratories, a trusted Contract Research Organization (CRO) with expertise in custom synthesis and analytical solutions, provides comprehensive support for Impurity Profiling and Characterization for Generic Project, helping pharmaceutical companies achieve regulatory compliance and successful approvals.
Article Highlights
- Impurity profiling and characterization are essential for generic drug submissions to the USFDA to ensure product safety, quality, and regulatory compliance.
- Pharmaceutical impurities can originate from synthesis processes, raw materials, residual solvents, or degradation, and must be properly identified, quantified, and controlled.
- Regulatory guidelines such as ICH Q3A and ICH Q3B define strict thresholds for impurity reporting, identification, and qualification during generic drug development.
- Advanced analytical techniques including HPLC, GC-MS, LC-MS/MS, and NMR are critical for accurate impurity detection, structural characterization, and stability evaluation.
- Proper impurity profiling helps demonstrate equivalence between generic drugs and reference listed drugs, supporting regulatory approval and long-term product quality.
- Comprehensive impurity management, including lifecycle monitoring and regulatory documentation, ensures consistent compliance and protects patient safety.
Understanding Impurity Profiling and Characterization
Impurity profiling involves the identification, quantification, and control of impurities present in a pharmaceutical product. This process is essential for ensuring the safety and efficacy of the drug. The USFDA has established guidelines for impurity profiling, including requirements for reporting, identifying, and qualifying impurities.
Importance of Impurity Profiling in Generic Drug Equivalence
Impurity profiling plays a vital role in demonstrating that a generic drug is comparable to its reference listed drug (RLD) in terms of safety and quality. While generic drugs are expected to have the same active ingredient, differences in manufacturing processes can lead to variations in impurity profiles. Regulatory authorities closely review these differences to ensure that no new or higher-risk impurities are introduced. A well-documented impurity profile provides scientific evidence that the generic product does not pose additional risks compared to the original product. This assessment is essential for gaining regulatory confidence and approval.
In addition, impurity profiling supports the overall evaluation of product consistency and manufacturing reliability. Consistent impurity levels across different production batches indicate that the manufacturing process is well controlled and reproducible. This helps establish trust in the long-term quality of the generic medicine. Without proper impurity evaluation, even minor process variations could go unnoticed and potentially affect product safety. Therefore, impurity profiling is not just a regulatory requirement but also a key component of quality assurance.
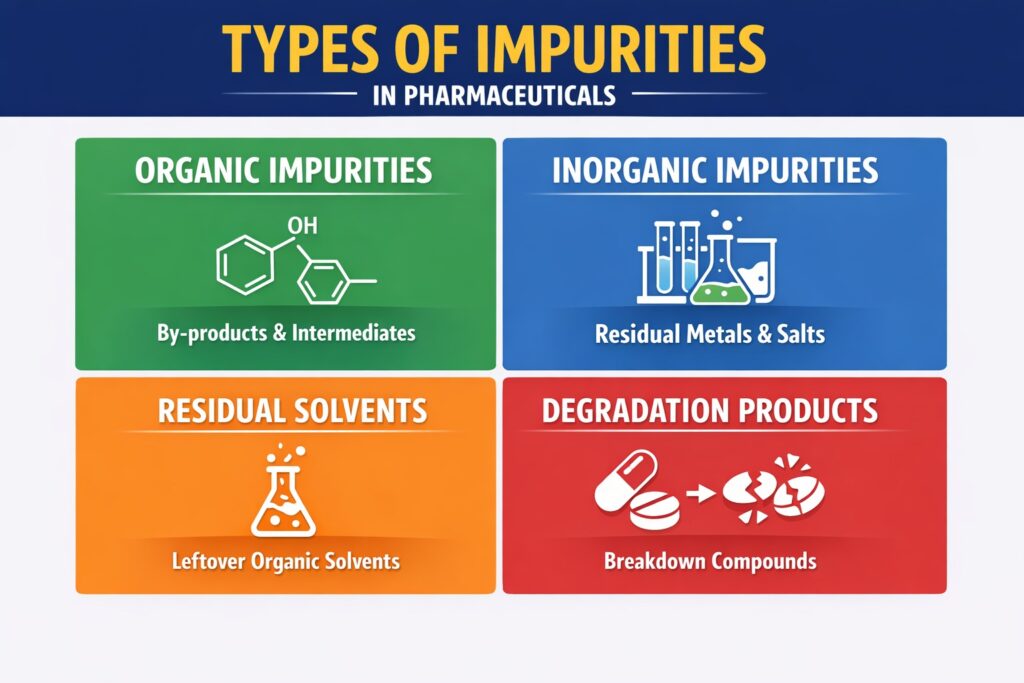
Types of Impurities
- Organic Impurities: These can arise during the synthesis of the drug substance and may include starting materials, intermediates, and by-products.
- Inorganic Impurities: These are often residual catalysts, reagents, or salts used in the manufacturing process.
- Residual Solvents: Organic solvents used in the synthesis process must be controlled to acceptable levels.
- Degradation Products: Impurities formed due to the degradation of the drug substance or product over time.
Regulatory Requirements
The USFDA’s guidelines for impurity profiling in generic drugs are detailed in documents such as ICH Q3A (R2) for drug substances and ICH Q3B (R2) for drug products. Key requirements include:
- Reporting Thresholds: Impurities must be reported if they are present above a certain threshold, typically 0.1% for drug substances and 0.2% for drug products.
- Identification Thresholds: Impurities above a specified level must be identified. This is usually 0.1% for drug substances and drug products.
- Qualification Thresholds: Impurities present above the qualification threshold must be evaluated for safety. This involves toxicological studies to determine their impact on human health.
Role of Forced Degradation Studies in Impurity Identification
Forced degradation studies are conducted to understand how a drug substance behaves under stress conditions such as heat, light, humidity, oxidation, and acidic or alkaline environments. These studies help identify potential degradation products that may form during manufacturing, storage, or handling. By intentionally accelerating the degradation process, scientists can predict long-term stability risks and develop appropriate control strategies. This information is essential for establishing stability-indicating analytical methods required for regulatory submissions.
Furthermore, forced degradation studies help ensure that analytical methods are capable of separating and detecting all relevant impurities. This confirms that the testing method is suitable for monitoring product quality throughout its shelf life. Regulatory agencies expect manufacturers to demonstrate that their analytical procedures can detect degradation products accurately. These studies also support packaging selection and storage condition recommendations. As a result, forced degradation testing plays a critical role in ensuring product safety and regulatory compliance.
Advanced Analytical Techniques for Impurity Profiling
At Resolvemass Laboratories, we employ state-of-the-art analytical techniques to provide comprehensive impurity profiling and characterization services. Our expertise in analytical chemistry and advanced instrumentation ensures accurate and reliable results.
Chromatography is a cornerstone of impurity profiling, enabling the separation and quantification of impurities in complex mixtures.
- High-Performance Liquid Chromatography (HPLC): HPLC is widely used for separating and quantifying organic impurities, including degradation products. Our advanced HPLC systems offer high resolution and sensitivity, ensuring precise impurity profiling.
- Gas Chromatography (GC): GC is used for volatile and semi-volatile impurities, including residual solvents. Coupled with mass spectrometry (GC-MS), this technique provides high sensitivity and specificity for impurity identification.
Spectroscopy techniques are essential for the structural elucidation and identification of impurities.
- Mass Spectrometry (MS): MS is a powerful tool for identifying and quantifying impurities with high sensitivity and specificity. Techniques such as Liquid Chromatography-Mass Spectrometry (LC-MS) and GC-MS are routinely used for impurity profiling.
- Nuclear Magnetic Resonance (NMR) Spectroscopy: NMR provides detailed structural information about impurities, aiding in their identification and characterization.
- Fourier-Transform Infrared (FTIR) Spectroscopy: FTIR is used to identify functional groups in impurities, complementing other spectroscopic techniques.
Hyphenated techniques combine chromatography and spectroscopy to provide comprehensive impurity profiling.
- LC-MS/MS: Liquid Chromatography coupled with tandem Mass Spectrometry (LC-MS/MS) offers high sensitivity and selectivity for detecting and quantifying impurities, particularly in complex matrices.
- GC-MS/MS: Gas Chromatography coupled with tandem Mass Spectrometry (GC-MS/MS) is ideal for volatile and semi-volatile impurities, providing high-resolution separation and accurate mass identification.
Importance of Reference Standards in Impurity Analysis
Reference standards are essential tools used to identify and quantify impurities with accuracy and confidence. These standards serve as comparison materials that help confirm the identity of unknown impurities detected during analysis. By comparing analytical responses such as retention time and mass spectra, scientists can verify the presence of specific impurity compounds. This ensures reliable impurity characterization and reduces uncertainty in analytical results.
In addition, qualified reference standards are required to support regulatory submissions and method validation. They help establish calibration curves and ensure that impurity levels are measured correctly. Without proper reference materials, it would be difficult to confirm the exact concentration and identity of impurities. Pharmaceutical companies often synthesize impurity standards when commercial sources are not available. This process strengthens regulatory documentation and improves overall analytical accuracy.
Our Comprehensive Impurity Profiling Services
Resolvemass Laboratories offers a full suite of impurity profiling and characterization services tailored to the requirements of generic drug submissions to the USFDA. Our services include:
1. Method Development and Validation
We develop and validate robust analytical methods for impurity profiling, ensuring compliance with USFDA guidelines. Our method validation process includes:
- Accuracy and Precision: Ensuring that the method produces reliable and reproducible results.
- Sensitivity and Specificity: Demonstrating the method’s ability to detect and quantify impurities at the required levels.
- Linearity and Range: Establishing the method’s ability to produce accurate results over a specified concentration range.
- Robustness: Assessing the method’s reliability under varying conditions.
We perform routine impurity testing for drug substances and drug products, providing detailed reports on the levels of impurities present. Our testing services include:
- Identification and Quantification: Identifying and quantifying impurities using advanced analytical techniques.
- Comparative Profiling: Comparing impurity profiles of generic drugs with their reference listed drugs (RLDs) to ensure bioequivalence.
- Stability Testing: Monitoring impurities over the shelf life of the product to assess its stability and compliance with regulatory standards.
3. Impurity Isolation and Characterization
For impurities that require further characterization, we offer isolation and structural elucidation services. This includes:
- Isolation: Using preparative chromatography to isolate impurities from the bulk drug substance or product.
- Structural Elucidation: Employing spectroscopic techniques such as NMR, MS, and FTIR to determine the chemical structure of impurities.
4. Regulatory Support and Documentation
We provide comprehensive support for regulatory submissions, including:
- Impurity Documentation: Preparing detailed reports and documentation on impurity profiling, including method validation data and impurity identification results.
- Regulatory Consultation: Offering expert guidance on USFDA requirements and strategies for impurity control and reporting.
Lifecycle Management of Impurity Profiles
Impurity profiling is not limited to the initial drug approval stage but continues throughout the entire product lifecycle. Changes in manufacturing processes, raw material suppliers, or production scale can affect impurity levels. Therefore, ongoing monitoring is necessary to ensure that impurity profiles remain within acceptable limits. Continuous evaluation helps manufacturers detect potential quality issues early and take corrective actions before they impact the market.
Lifecycle impurity management also supports post-approval regulatory compliance. Pharmaceutical companies must assess the impact of any process change on impurity formation and report significant findings to regulatory authorities. This ensures transparency and maintains product approval status. Regular review of impurity trends helps maintain consistent product quality. It also demonstrates a strong commitment to patient safety and regulatory responsibility.
Case Study: impurity-profiling-and-characterization-for-generic-project
A pharmaceutical company approached Resolvemass Laboratories for impurity profiling services for their generic drug submission to the USFDA. The project involved:
Challenges
- Complex Formulation: The generic drug had a complex formulation, requiring advanced analytical techniques for impurity profiling.
- Low-Level Impurities: The presence of low-level impurities necessitated highly sensitive and specific analytical methods.
- Regulatory Compliance: The company needed to ensure compliance with USFDA guidelines for impurity reporting, identification, and qualification.
Solution
- Method Development and Validation: We developed and validated a robust HPLC method for impurity profiling, ensuring high sensitivity and specificity.
- Advanced Analytical Techniques: We employed LC-MS/MS and NMR spectroscopy for the identification and quantification of impurities, including trace-level degradation products.
- Comprehensive Reporting: We provided the client with detailed reports and regulatory documentation, including impurity profiles, method validation data, and safety assessments.
Outcome
The successful impurity profiling enabled the pharmaceutical company to meet USFDA requirements and submit a complete and compliant regulatory dossier. Our comprehensive testing and regulatory support facilitated the approval process, ensuring the timely market entry of the generic drug.
Common Challenges in Impurity Profiling for Generic Drug Submissions
One of the major challenges in impurity profiling is detecting impurities present at extremely low concentrations. These trace impurities require highly sensitive analytical instruments and optimized methods to ensure accurate detection. In some cases, impurities may co-elute with the main drug compound, making separation difficult. This requires advanced method development strategies and careful optimization of analytical conditions.
Another challenge involves identifying unknown impurities that do not have available reference standards. Structural characterization of these impurities requires a combination of advanced analytical techniques and scientific expertise. Additionally, tight regulatory timelines can create pressure to complete impurity studies quickly without compromising data quality. Addressing these challenges requires experienced analytical teams and modern instrumentation to ensure reliable and compliant results.
Conclusion
Impurity profiling and characterization are critical for ensuring the safety, efficacy, and quality of generic drugs. At Resolvemass Laboratories, we offer advanced impurity profiling services that meet stringent USFDA guidelines, supporting successful generic drug submissions. Our commitment to quality, precision, and regulatory compliance makes us a trusted partner for pharmaceutical companies seeking to navigate the complex regulatory landscape and bring high-quality generic drugs to market.
In addition, impurity profiling supports consistent product quality and helps manufacturers maintain control over their processes throughout the product lifecycle. Continuous monitoring ensures that impurity levels remain within safe and acceptable limits, even when manufacturing changes occur. This proactive approach helps prevent regulatory issues and protects patient safety. By investing in robust impurity characterization and expert analytical support, pharmaceutical companies can improve approval success and confidently deliver safe, high-quality generic medicines to the market.
Reference:
- Ahuja, S., & Alsante, K. M. (2003). Overview: Isolation and characterization of impurities. In Handbook of isolation and characterization of impurities in pharmaceuticals (Vol. 5). Academic Press. https://www.sciencedirect.com/science/article/pii/B9780123756800000140
- Ahuja, S. (2003). Overview: Isolation and characterization of impurities. In S. Ahuja (Ed.), Handbook of isolation and characterization of impurities in pharmaceuticals (Progress in Pharmaceutical and Biomedical Analysis, Vol. 5). Academic Press. https://doi.org/10.1016/S0149-6395(03)80003-X
- Kruhlak, N. L., Benz, R. D., Zhou, H., Colatsky, T. J., & Richardson, J. S. (2007). Progress in QSAR toxicity screening of pharmaceutical impurities. Advanced Drug Delivery Reviews, 59(1), 77–89. https://doi.org/10.1016/j.addr.2006.10.002
GET IN TOUCH WITH US
For more information about our impurity profiling and characterization services and how we can assist with your generic drug submission to the USFDA, please Resolvemass laboratories

CMC Strategy for Leuprolide Depot ANDA Submission
Introduction: Why Leuprolide Depot CMC Strategy Is Among the Most Complex in the ANDA World…

Step-by-Step Guide to Enzymatic Digestion Strategies for GLP-1 Peptide Mapping
Introduction: GLP-1 Enzymatic Digestion Mapping is a structured analytical process where enzymes are used to…

Why Peptide Mapping is Critical for GLP-1 Analog Characterization in Generic Development
Introduction to GLP-1 Peptide Characterization Mapping GLP-1 Peptide Characterization Mapping is an essential scientific approach…

How to Perform Peptide Mapping of GLP-1 Analogs Using LC-MS/MS
Introduction: GLP-1 Peptide Mapping Using LC-MS is a powerful analytical approach used to confirm sequence…

How to Develop PLGA Microsphere-Based Injectables
Introduction: The development of long-acting injectables (LAIs) relies on careful design and optimization of poly(lactic-co-glycolic…

Peptide Mapping of GLP-1 Peptides: Complete Analytical Workflow for Structural Characterization
Introduction: Peptide Mapping GLP-1 Peptides is a fundamental analytical strategy used in the structural characterization…

