
Introduction:
Matrix effects in LC-MS/MS bioanalysis are among the most significant challenges affecting the reliability of pharmacokinetic, toxicokinetic, and therapeutic drug monitoring data. Instead of being regarded as a minor background interference, this phenomenon involves substantial changes in the ionization efficiency of target analytes caused by co-eluting endogenous or exogenous compounds within the mass spectrometer ion source. In today’s high-throughput analytical laboratories, the presence of undetected interfering substances can produce unpredictable ion suppression or ion enhancement, ultimately compromising assay accuracy, precision, and the lower limit of quantification (LLOQ). Establishing a robust and regulatory-compliant bioanalytical method demands a comprehensive understanding of the physicochemical processes responsible for these matrix effects, ranging from alterations in droplet surface properties within the electrospray plume to competition during gas-phase proton transfer. This article presents an in-depth examination of the underlying causes of ionization interference, validated approaches for quantitative assessment, current international regulatory expectations, and advanced mitigation techniques that align with the rigorous scientific standards required for successful regulatory submissions.
Develop a Flawless Analytical Framework: Uncover how strategic planning safeguards your data against matrix interference by exploring our Bioanalytical Strategy for Drug Development.
Share via:
Article Summary:
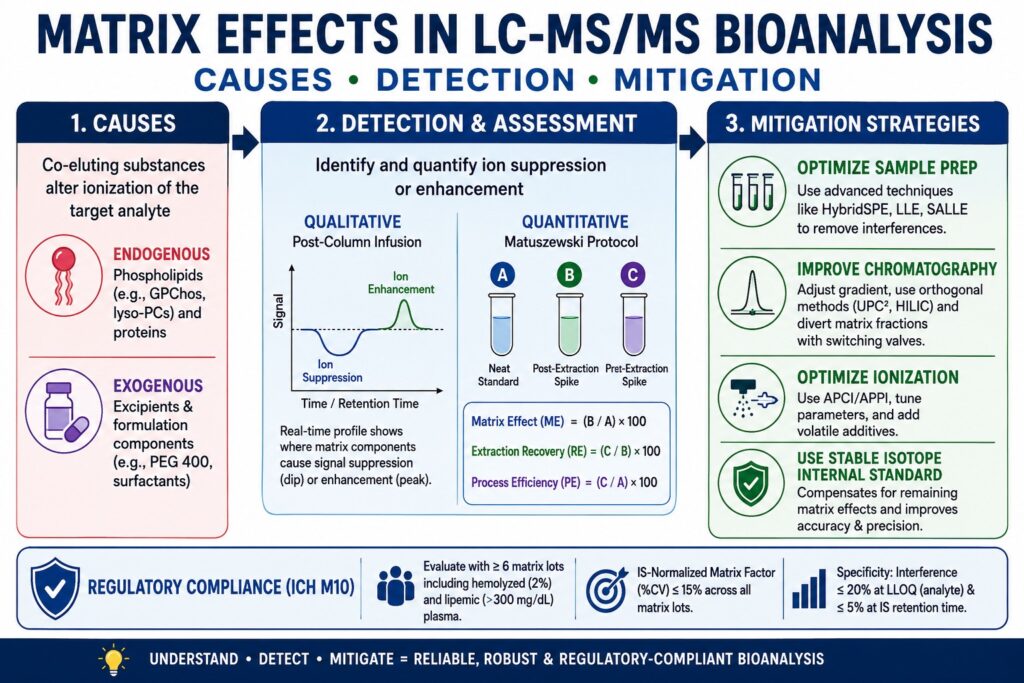
- Matrix effects in LC-MS/MS occur when endogenous biological molecules or exogenous substances alter analyte ionization during mass spectrometry, leading to ion suppression or ion enhancement that can compromise assay accuracy, precision, sensitivity, and quantification.
- Major sources of interference include endogenous phospholipids, proteins, and salts, as well as exogenous compounds such as formulation excipients (e.g., PEG 400, Tween 80), anticoagulants, and co-administered drugs that compete with analytes during the ionization process.
- Reliable detection and measurement of matrix effects rely on complementary approaches such as post-column infusion for identifying suppression or enhancement regions and the Matuszewski post-extraction addition protocol for calculating matrix effect, extraction recovery, and overall process efficiency.
- Regulatory guidelines, including ICH M10, require comprehensive matrix effect evaluation using multiple independent matrix lots, assessment in hemolyzed and lipemic samples, and demonstration that the IS-normalized matrix factor remains within acceptable precision limits to ensure method robustness.
- Reducing matrix interference begins with effective sample preparation strategies, including liquid-liquid extraction (LLE), solid-phase extraction (SPE), HybridSPE, and optimized cleanup procedures that selectively remove phospholipids and other interfering matrix components before analysis.
- Additional mitigation techniques include optimizing chromatographic separation, selecting appropriate ionization sources such as APCI or APPI when suitable, fine-tuning instrument parameters, and using stable isotope-labeled internal standards (SIL-IS) to compensate for residual ionization variability.
- A comprehensive mitigation strategy that integrates optimized extraction, chromatographic optimization, quantitative matrix assessment, regulatory-compliant validation, and appropriate internal standards produces robust, reproducible, and regulatory-ready LC-MS/MS bioanalytical methods across diverse biological matrices.
Primary Causes of Matrix Effects in LC-MS/MS Bioanalysis
The principal causes of matrix effects in LC-MS/MS bioanalysis arise from co-eluting endogenous biological constituents, including phospholipids, as well as exogenous substances such as formulation excipients. These compounds compete directly with the target analyte for available charge during the atmospheric pressure ionization process, resulting in altered ionization efficiency. Identifying both the origin and the physicochemical mechanism responsible for this interference is an essential step in developing a reliable, reproducible, and validated bioanalytical method.
Endogenous Biological Interferences: Phospholipids and Proteins
Endogenous phospholipids, particularly glycerophosphocholines (GPChos) and lysophosphatidylcholines (lyso-PCs), are widely recognized as the most prevalent and problematic contributors to ion suppression in plasma, serum, and tissue-based matrices. These amphiphilic molecules are naturally present at very high concentrations, approximately 1 mg/mL in human plasma, and they readily co-extract with numerous analytes during conventional protein precipitation (PPT) and standard solid-phase extraction (SPE) procedures. Their simultaneous extraction is primarily attributed to their structural characteristics, consisting of a hydrophobic lipid tail combined with a highly polar zwitterionic head group.
During Electrospray Ionization (ESI), the mechanism responsible for ion suppression becomes particularly evident within the Taylor cone. As the applied high-voltage electric field generates a fine spray of highly charged droplets, amphiphilic matrix components accumulate preferentially at the droplet surface. According to both the Ion Evaporation Model and the Charge Residue Model, this surface accumulation increases the surface tension and viscosity of the droplets, significantly reducing solvent evaporation efficiency and limiting the successful transfer of target analytes into the gas phase. In addition to these physical effects, compounds possessing high gas-phase basicity may further reduce analyte response by neutralizing already-ionized analytes through secondary proton transfer reactions occurring in the gas phase.
Overcome Complex Extraction Challenges: Learn how specialized methods isolate analytes from complex biological environments in our Tissue and CSF Bioanalytical Services.
During method development, phospholipids can be systematically monitored and characterized by scanning for their characteristic mass-to-charge (m/z) transitions:
- Glycerophosphocholines (GPChos): Produce a characteristic product ion at m/z 184, corresponding to the cleaved phosphocholine polar head group.
- Lysophosphatidylcholines (2-lyso GPChos): Predominantly generate fragment ions at m/z 104.
The relatively late elution of intact GPChos on reversed-phase (RP) chromatographic columns creates an additional analytical concern. When the high-organic column wash step is not sufficiently extended, residual phospholipids may continue to elute during subsequent sample injections. This carryover results in unpredictable and continuously shifting ion suppression, often referred to as “rolling” ion suppression, affecting multiple injections throughout an analytical batch.
Exogenous Interferences: Dosing Vehicles, Excipients, and Co-Medications
Exogenous matrix effects arise from substances intentionally or unintentionally introduced into the biological system or analytical sample. Common sources include pharmaceutical formulation excipients, anticoagulants, sample stabilization buffers, and concurrently administered medications. During preclinical studies, formulations frequently contain elevated concentrations of solubilizing agents such as Polyethylene Glycol 400 (PEG 400), Tween 80 (polysorbate), and propylene glycol. These compounds can remain in systemic circulation and subsequently appear at significant concentrations within collected plasma samples.
Among these excipients, PEG 400 is particularly problematic because it produces a broad and highly intense chromatographic peak consisting of multiple oligomeric species, including masses of 327, 371, 414, 458, 502, and 546, each separated by repeating units of 44 Da. When PEG 400 co-elutes with early- or mid-eluting basic drug compounds during reversed-phase chromatography, it dominates the ionization process by consuming a substantial proportion of the available ionization energy within the source. The resulting competition produces pronounced ion suppression of the target analyte. Published pharmacokinetic screening studies have demonstrated that the presence of PEG 400 can artificially reduce measured plasma concentrations of specific analytes, leading to an overestimation of plasma clearance values by approximately two- to five-fold. Such analytical distortion may result in the premature elimination of otherwise promising drug candidates during early drug discovery programs, emphasizing the importance of monitoring formulation excipients together with the analyte throughout method development and validation.
Accelerate Preclinical Outcomes: Discover how to identify and bypass early formulation interferences with our Bioanalytical Services for Rapid Proof-of-Concept.
Detection and Quantitative Assessment of Matrix Effects in LC-MS/MS Bioanalysis
Matrix effects are primarily detected and quantitatively evaluated using two complementary techniques: post-column infusion for continuous qualitative visualization and the Matuszewski post-extraction addition protocol for accurate mathematical determination of the matrix factor. Proper implementation of these assessment strategies ensures that concealed regions of ion suppression or enhancement are identified, measured, and effectively addressed before formal bioanalytical method validation begins.
Post-Column Infusion for Qualitative Profiling
Post-column infusion is widely used to generate a real-time qualitative profile of ion suppression and ion enhancement throughout the complete chromatographic run, allowing analysts to precisely identify where interfering matrix components elute. In this experimental configuration, a continuous and stable flow of the pure target analyte together with its internal standard is introduced into the liquid chromatography effluent through a T-union immediately before the stream enters the mass spectrometer ion source. At the same time, an extracted blank biological matrix is injected onto the analytical column.
Because the mass spectrometer continuously monitors the analyte-specific Multiple Reaction Monitoring (MRM) transition, the signal baseline is expected to remain stable under ideal conditions. However, as endogenous or exogenous matrix constituents elute from the chromatographic column and enter the ion source, they alter the ionization efficiency of the continuously infused analyte. A decrease in signal intensity appears as a depression or dip in the baseline, indicating regions of ion suppression, whereas an increase in signal intensity produces elevated peaks that represent ion enhancement. By comparing the retention time of the target analyte with the resulting infusion chromatogram, analytical scientists can immediately determine whether the analyte elutes within a high-risk region populated by otherwise undetectable matrix interferents. This information provides critical guidance on whether chromatographic conditions require further optimization to eliminate or minimize matrix-induced ionization effects.
The Matuszewski Post-Extraction Addition Protocol
The Matuszewski post-extraction addition protocol is specifically designed to quantitatively distinguish the influence of matrix-induced ionization effects from the physical loss of analyte that occurs during the extraction process. First introduced in 2003, this approach continues to be regarded as the gold standard for the quantitative evaluation of matrix effects. The procedure requires the preparation and comparison of three separate sample sets, each serving a distinct analytical purpose:
Set A (Neat Standard): The analyte is spiked directly into the reconstitution solvent or mobile phase without the presence of any biological matrix. This sample represents the theoretical condition of 100% ionization efficiency and serves as the reference response free from matrix interference.
Set B (Post-Extraction Spike): A blank biological matrix is first subjected to the intended sample preparation and extraction procedure. After extraction is complete, the analyte is added to the final extract immediately before instrumental analysis. This sample isolates the effect of the biological matrix on ionization while excluding analyte loss during extraction.
Set C (Pre-Extraction Spike): The analyte is added to the blank biological matrix before any extraction steps are performed. This preparation closely replicates the behavior of an authentic study sample because the analyte experiences both the extraction process and any subsequent matrix-induced ionization effects.

Using the absolute peak area responses generated from these three sample sets, bioanalytical scientists calculate three essential performance parameters that collectively evaluate the analytical method:
| Performance Parameter | Calculation Formula | Scientific Interpretation |
|---|---|---|
| Matrix Effect (ME) | ME (%) = (B/A) × 100 | Determines the extent of ionization alteration caused by the biological matrix. ME values below 100% indicate ion suppression, whereas values above 100% indicate ion enhancement. |
| Extraction Recovery (RE) | RE (%) = (C/B) × 100 | Measures the actual efficiency of the chemical extraction process while remaining completely independent of ionization effects. |
| Process Efficiency (PE) | PE (%) = (C/A) × 100 | Represents the combined overall influence of analyte recovery during extraction together with matrix-induced changes in ionization efficiency. |
For reliable quantitative analysis, assay performance is not determined solely by the absolute matrix effect, which reflects deviation from the ideal value of 100%. Equally important is the relative matrix effect, which represents the variability of the absolute matrix effect across multiple independent sources of the biological matrix and is expressed as the coefficient of variation (%CV). Excessive variability in the relative matrix effect directly translates into increased variability in calibration curve slopes, reducing assay reproducibility and limiting the reliability of quantitative results across large and diverse patient populations.
Mitigate Incurred Sample Variances: Ensure your assay remains robust during post-extraction phases by understanding Incurred Sample Reanalysis (ISR) in Bioanalytical Studies.
Slope Ratio Analysis and Matrix-Matched Calibration
Slope Ratio Analysis serves as either an alternative or a complementary approach to the post-extraction addition method by evaluating matrix effects throughout the entire linear dynamic range instead of at isolated concentration levels. This strategy requires the preparation of two complete calibration curves: one generated using a neat standard solution and another prepared using a matrix-matched post-extraction sample.
The proportional influence of the biological matrix is assessed by comparing the slopes of the resulting linear regression equations.
Signal Suppression/Enhancement (SSE %) = (Slopematrix-matched / Slopeneat standard) × 100
When the slope obtained from the matrix-matched calibration curve is lower than that of the neat standard calibration curve, the result confirms proportional ion suppression across the calibration range. Conversely, a higher slope indicates ion enhancement. This analytical approach is particularly valuable for identifying concentration-dependent matrix effects, which may introduce significant non-linearity, especially within the lower region of the calibration curve where accurate quantification is most challenging.
Regulatory Guidelines and Acceptance Criteria: ICH M10 Harmonization
Global regulatory authorities require comprehensive evaluation of matrix effects to protect patient safety and preserve the integrity of bioanalytical data. The ICH M10 guideline currently serves as the internationally harmonized standard adopted by regulatory agencies, including the FDA, EMA, and numerous other health authorities. According to ICH M10, every bioanalytical method must convincingly demonstrate that the selected internal standard (IS) effectively tracks and compensates for matrix-induced variability across a wide range of challenging biological matrices and subject populations.
IS-Normalized Matrix Factor Requirements
The principal quantitative parameter required by ICH M10 for chromatographic bioanalytical methods is the IS-Normalized Matrix Factor (IS-Normalized MF). This value is determined by dividing the absolute Matrix Factor of the target analyte by the corresponding absolute Matrix Factor of the internal standard. When the internal standard accurately mirrors the analyte throughout the ionization process and experiences an identical degree of ion suppression or ion enhancement, the resulting ratio is expected to equal 1.0.
During formal bioanalytical method validation, regulatory acceptance criteria are highly stringent. The precision of the IS-normalized matrix factor, expressed as the coefficient of variation (%CV) across all evaluated biological matrix lots, must not exceed 15%. By enforcing this acceptance limit of ≤15% CV, regulatory agencies ensure that relative matrix effects do not negatively impact assay precision when analyzing samples collected from hundreds of individual clinical trial participants, each possessing naturally different biological matrix compositions.
Navigate Global Compliance Standards: Learn how to implement these parameters smoothly across jurisdictions by reviewing the ICH M10 Bioanalytical Method Validation Guidelines.
Matrix Diversity and Special Populations
To demonstrate that a bioanalytical method remains reliable across diverse biological conditions, ICH M10 specifies detailed requirements regarding the selection of biological matrices during method validation.
Six-Lot Requirement: Matrix effect evaluation must be conducted using a minimum of six independent lots of blank biological matrix obtained from different individual donors. This requirement ensures that normal biological variability is adequately represented during validation.
Hemolyzed Matrix: Pre-analytical errors occurring during blood collection frequently result in red blood cell lysis. Consequently, validation studies must include at least one highly hemolyzed matrix lot, which is commonly prepared by adding 2% lysed whole blood to blank plasma. Hemolysis introduces substantial quantities of intracellular proteins together with iron-containing components that significantly alter ionization behavior within the mass spectrometer.
Lipemic Matrix: Biological variability associated with post-prandial sample collection often produces elevated circulating lipid concentrations. Therefore, validation must also include at least one highly lipemic matrix lot containing greater than 300 mg/dL of triglycerides. The increased concentration of complex lipids creates substantial competition during ionization and may significantly influence analyte response.
Relevant Patient Populations: When a therapeutic compound is specifically intended for patient populations exhibiting altered metabolic characteristics, such as individuals with renal failure or hepatic impairment, biological matrices collected directly from these patient groups should also be incorporated into method validation. This additional evaluation helps confirm that the analytical method maintains acceptable performance during Phase II and Phase III clinical studies under real-world biological conditions.
Matrix effect assessment must be performed using both Low Quality Control (LQC) and High Quality Control (HQC) concentrations, with a minimum of three replicate analyses conducted for each biological matrix lot. For every individual matrix lot evaluated during validation, the accuracy of the back-calculated concentration must remain within ±15% of the corresponding nominal concentration, thereby demonstrating consistent analytical performance across all tested biological matrices.
Streamline Late-Stage Asset Audits: Secure flawless transitions to larger target cohorts with our Bioanalytical CRO Services for Phase II & Phase III.
Specificity and Selectivity Thresholds
Although the Matrix Factor is used to evaluate changes in ionization efficiency, regulatory guidelines also establish strict acceptance criteria for direct baseline interference originating from matrix components that generate the same mass transitions as the target analyte. According to the ICH M10 guideline, the signal produced by any interfering compound present in a blank biological matrix at the retention time corresponding to the analyte must not exceed 20% of the mean analyte response measured at the lower limit of quantification (LLOQ). Likewise, any interference detected at the retention time of the internal standard must remain ≤ 5% of the corresponding internal standard (IS) response. These stringent selectivity requirements ensure that endogenous or exogenous matrix constituents do not compromise accurate identification or reliable quantification of either the analyte or the internal standard.
Resolve Auditing Roadblocks Early: Understand the key indicators that lead to regulatory dynamic shortfalls in our guide on Bioanalytical Method Validation Failure.
Advanced Mitigation Strategies for Matrix Effects in LC-MS/MS Bioanalysis
Mitigating matrix effects in LC-MS/MS bioanalysis requires a comprehensive, multi-layered strategy that integrates optimized sample preparation, orthogonal chromatographic separation, alternative ionization techniques, and the appropriate use of stable isotope-labeled internal standards. Since no single approach is capable of eliminating matrix effects under every analytical condition, bioanalytical scientists typically combine several complementary techniques to achieve consistent assay performance that satisfies regulatory expectations.
Optimization of Sample Preparation Protocols
The most direct and effective strategy for minimizing matrix effects is to physically separate the target analyte from interfering biological components before the sample enters the liquid chromatography system. The extraction technique selected during method development has a substantial impact on the cleanliness of the final extract and, consequently, on the degree of ion suppression or ion enhancement observed during analysis.
| Sample Preparation Technique | Mechanism | Impact on Matrix Effects |
|---|---|---|
| Protein Precipitation (PPT) | Organic solvents such as acetonitrile or methanol denature proteins, causing them to precipitate from solution. | Poor. More than 95% of soluble phospholipids, salts, and formulation excipients remain in the extract, frequently resulting in substantial ion suppression. |
| Liquid-Liquid Extraction (LLE) | Analytes partition into an immiscible organic solvent through pH-dependent charge neutralization. | Good. Highly polar salts, proteins, and strongly zwitterionic phospholipids generally remain within the aqueous phase, reducing matrix interference. |
| Salting-Out Assisted LLE (SALLE) | High salt concentrations induce phase separation of water-miscible organic solvents, such as acetonitrile, into a distinct organic layer. | Very Good. This approach is broadly suitable for numerous lipophilic analytes while producing cleaner extracts than conventional protein precipitation. |
| Mixed-Mode Solid Phase Extraction (SPE) | Combines reversed-phase retention with ion-exchange mechanisms, such as strong cation exchange, to selectively retain analytes. | Excellent. Charged analytes are efficiently retained while neutral lipids, PEG 400, and other interfering compounds can be selectively removed during washing steps. |
| HybridSPE (Phospholipid Removal) | Employs zirconia-coated sorbents or Lewis acid-base interactions to selectively bind phosphate-containing molecules. | Exceptional. Specifically captures and removes phospholipids while allowing analytes lacking phosphate groups to pass through with minimal interference, effectively eliminating one of the primary causes of biological ion suppression. |
When simple Protein Precipitation (PPT) must be employed because of budget limitations or high-throughput analytical requirements, substantial dilution of the final extract, typically between 5-fold and 10-fold, is often necessary. Dilution proportionally decreases the concentration of competing matrix constituents entering the mass spectrometer ion source, frequently reducing their abundance below the saturation threshold and thereby restoring the ionization efficiency of the target analyte.
Align with Best Industry Practices: See how structural sample prep selection changes based on your outsourcing model via our Bioanalytical CRO Partnership framework.
Chromatographic Adjustments and Orthogonal Separations
When chemical extraction alone cannot sufficiently eliminate interfering matrix constituents, optimization of high-performance liquid chromatography (HPLC) conditions becomes essential. The primary objective is to alter chromatographic retention so that the analyte elutes separately from matrix-derived interferents, thereby minimizing competition during ionization.
Mobile Phase and Gradient Adjustments: Modifying the mobile phase gradient by making it shallower can significantly improve the chromatographic resolution of closely eluting compounds. Because lysophosphatidylcholines generally elute earlier than intact phosphatidylcholines on reversed-phase chromatographic columns, changing the organic solvent, for example replacing methanol with acetonitrile or isopropanol, can selectively alter the retention behavior of these lipid classes. Such adjustments help separate lipid-derived matrix components from the analyte of interest, thereby reducing matrix-induced ionization effects.
Orthogonal Chromatography: For highly hydrophobic analytes that naturally co-elute with late-eluting phospholipids under conventional reversed-phase conditions, adopting orthogonal chromatographic techniques provides a highly effective solution. UltraPerformance Convergence Chromatography (UPC2), which utilizes supercritical carbon dioxide as the principal mobile phase, offers markedly different selectivity compared with reversed-phase chromatography. In practical bioanalytical applications, UPC2 has successfully separated highly hydrophobic compounds, including clopidogrel, from lipid-based interferences without relying on highly sophisticated extraction procedures. Similarly, Hydrophilic Interaction Liquid Chromatography (HILIC) offers significant advantages for highly polar analytes by providing alternative retention mechanisms that improve separation from interfering biological matrix components.
Column Flushing and Switching Valves: The incorporation of automated post-column switching valves provides an additional strategy for minimizing matrix contamination. During the initial phase of the chromatographic run, fractions containing unretained salts and highly polar interferents can be diverted directly to waste. Likewise, the high-organic wash phase at the end of the gradient, which typically contains late-eluting phospholipids and other strongly retained contaminants, can also be excluded from entering the mass spectrometer. Preventing these high matrix loads from reaching the ion source significantly reduces contamination while improving long-term instrument stability and analytical reproducibility.
Ionization Source Modification and Instrument Parameter Tuning
The extent of matrix effects is strongly influenced by the ionization mechanism employed during LC-MS/MS analysis. Electrospray Ionization (ESI) is particularly susceptible to ion suppression because ion formation occurs within charged liquid droplets, where factors such as surface tension, droplet composition, and localized microenvironments directly influence the efficiency of charge transfer and analyte ionization.
Transitioning from ESI to APCI or APPI
Atmospheric Pressure Chemical Ionization (APCI) and Atmospheric Pressure Photoionization (APPI) generate ions primarily in the gas phase rather than within liquid droplets, making them considerably less vulnerable to matrix-induced ionization effects. During APCI, the liquid chromatography effluent is completely vaporized using elevated temperatures before ionization occurs through a corona discharge needle. Since desolvation is driven predominantly by thermal energy instead of liquid-phase surface phenomena, APCI and APPI exhibit substantially greater resistance to matrix-related ion suppression. Although APCI may produce lower absolute signal intensity than ESI when analyzing highly polar or thermally sensitive compounds, the substantial reduction in relative matrix variability frequently makes APCI the preferred ionization technique for analytical methods requiring compliance with stringent regulatory precision criteria.
Tuning Instrument Parameters
Optimization of instrument operating parameters can significantly reduce matrix effects and improve analytical performance. The capillary or sprayer voltage directly influences ionization efficiency and should be carefully optimized during method development. Lower liquid chromatography flow rates, including micro-LC and nano-LC configurations, generate smaller initial droplets within the Electrospray Ionization source. These smaller droplets require less energy to achieve complete desolvation while simultaneously reducing the concentration of competing matrix constituents within each droplet, thereby improving analyte ionization efficiency.
In addition, the incorporation of volatile mobile phase additives such as ammonium acetate or formic acid, together with suitable dopants including isopropanol, can reduce droplet surface tension and shift the ionization equilibrium in favor of the target analyte. These adjustments help counteract the detrimental influence of endogenous salts and other competing matrix components, resulting in more stable ionization and improved analytical robustness.
Account for Analyte Thermal Deviations: Ensure matrix parameters remain robust over extended analytical timelines by checking our insights on Stability Testing in Bioanalysis.
Strategic Implementation of Stable Isotope-Labeled Internal Standards (SIL-IS)
When optimized sample preparation and instrumental strategies are insufficient to completely eliminate matrix effects, the incorporation of a Stable Isotope-Labeled Internal Standard (SIL-IS) serves as the most reliable analytical safeguard for achieving accurate quantitative results. A SIL-IS is generally synthesized by replacing selected atoms within the target analyte with stable heavy isotopes such as Carbon-13 (¹³C), Nitrogen-15 (¹⁵N), or Deuterium (²H) while preserving the compound’s original chemical identity.
Because the SIL-IS possesses the same chemical structure, pKa, hydrophobicity, and overall physicochemical characteristics as the native analyte, it behaves almost identically throughout the analytical workflow. It undergoes the same extraction process, exhibits identical chromatographic retention, and co-elutes precisely with the target analyte during liquid chromatography. As a result, both the analyte and the SIL-IS are exposed to the same extent of ion suppression or ion enhancement within the mass spectrometer ion source.
Quantitative analysis is performed by calculating the ratio between the analyte peak area and the internal standard peak area. Since both compounds experience nearly identical matrix-induced ionization effects, this ratio effectively compensates for the absolute matrix effect, thereby restoring both assay accuracy and precision. Nevertheless, an important limitation must be recognized. A SIL-IS cannot eliminate severe absolute ion suppression. If matrix interference suppresses approximately 99% of the analyte signal, the resulting signal-to-noise (S/N) ratio will remain insufficient to satisfy sensitivity requirements at the lower limit of quantification (LLOQ), even when the analyte-to-internal standard ratio maintains acceptable linearity. Consequently, a SIL-IS should never be considered a substitute for proper method development or optimized sample preparation. Instead, it should always be implemented alongside efficient sample cleanup procedures that minimize matrix interference before instrumental analysis.
In situations where a SIL-IS is either commercially unavailable or prohibitively expensive, structural analog internal standards are commonly selected as practical alternatives. However, these analog compounds require comprehensive validation because even relatively small differences in chromatographic retention, extraction behavior, or ionization efficiency can produce substantially different matrix effects compared with the target analyte. Such discrepancies may reduce compensation efficiency and ultimately compromise assay accuracy, precision, and overall method performance.
Tailor Assays for Complex Biologics: Discover advanced validation and tracking setups for challenging synthetic modalities through our specialized Oligonucleotide Bioanalytical Services.
Conclusion
Successfully controlling Matrix Effects in LC-MS/MS Bioanalysis requires a comprehensive scientific approach that combines a thorough understanding of biological matrix composition, the fundamental principles of mass spectrometry, and strict adherence to internationally accepted regulatory requirements. Matrix-induced ion suppression originating from abundant endogenous phospholipids, together with pronounced signal attenuation caused by exogenous substances such as preclinical formulation vehicles including PEG 400, has the potential to significantly distort quantitative bioanalytical results if left unrecognized. These interferences can adversely affect pharmacokinetic evaluations, toxicokinetic investigations, and therapeutic drug monitoring by introducing substantial inaccuracies into measured analyte concentrations.
A systematic strategy that integrates post-column infusion profiling, quantitative Matrix Factor determination using the Matuszewski post-extraction addition protocol, advanced sample preparation techniques such as HybridSPE, optimized chromatographic separation, and comprehensive validation according to ICH M10 requirements enables bioanalytical scientists to accurately identify, quantify, and effectively mitigate matrix-related variability. The thoughtful application of these complementary methodologies enhances assay robustness, improves reproducibility, and ensures reliable quantitative performance across diverse biological matrices and patient populations.
Developing expertise in these advanced mitigation strategies ultimately supports the generation of highly accurate, reproducible, and scientifically defensible LC-MS/MS bioanalytical data. Such rigor not only strengthens confidence in pharmacokinetic and clinical study outcomes but also ensures that analytical methods consistently satisfy the demanding expectations of global regulatory authorities throughout drug development and regulatory submission.
For specialized analytical support, rigorous method validation workflows, and advanced bioanalytical methodology consulting, visit the ResolveMass Laboratories Inc. portal: Contact Us
Frequently Asked Questions (FAQs)
Endogenous phospholipids are amphiphilic molecules that naturally accumulate at the surface of charged droplets during Electrospray Ionization (ESI). Their presence interferes with solvent evaporation and disrupts efficient charge transfer to the analyte. This competition limits the formation of analyte ions entering the gas phase, resulting in reduced signal intensity. Consequently, phospholipids are considered one of the most significant contributors to ion suppression in LC-MS/MS bioanalysis.
Atmospheric Pressure Chemical Ionization (APCI) performs ionization after the liquid mobile phase has been completely vaporized, meaning the process occurs predominantly in the gas phase. Since APCI does not depend on charged liquid droplets for ion formation, it is much less affected by droplet surface properties and charge competition. This characteristic makes APCI more resistant to matrix-induced ion suppression than Electrospray Ionization (ESI), particularly for suitable analytes.
According to the ICH M10 guideline, matrix effects must be evaluated using at least six independent lots of blank biological matrix collected from different donors. The Internal Standard (IS)-normalized Matrix Factor should be determined at both Low Quality Control (LQC) and High Quality Control (HQC) concentrations. The coefficient of variation (%CV) for the IS-normalized Matrix Factor across all evaluated lots must remain within 15%. These requirements help demonstrate consistent assay performance across diverse biological matrices.
A Stable Isotope-Labeled Internal Standard (SIL-IS) closely resembles the target analyte in chemical structure while containing stable heavy isotopes that distinguish it during mass spectrometric detection. Because both compounds co-elute and undergo identical extraction and ionization processes, they experience nearly the same matrix effects. Calculating the analyte-to-internal standard response ratio compensates for signal variations caused by the matrix, thereby improving quantitative accuracy and precision.
Yes. A symmetrical chromatographic peak only indicates satisfactory chromatographic separation and does not guarantee the absence of matrix effects. Ion suppression or enhancement occurs inside the mass spectrometer ion source, where co-eluting matrix components may alter analyte ionization without producing detectable chromatographic peaks. As a result, significant signal suppression can occur even when the chromatographic peak appears perfectly normal.
Formulation excipients such as Polyethylene Glycol 400 (PEG 400) and Tween 80 are frequently used to improve the solubility of poorly water-soluble drug candidates during preclinical studies. These substances may remain in circulation and appear in collected biological samples. During LC-MS/MS analysis, they can produce broad chromatographic signals and compete strongly for ionization within the mass spectrometer source. This competition may substantially suppress analyte signals and lead to inaccurate quantitative results if not properly controlled.
Diluting the final sample extract decreases the concentration of salts, phospholipids, proteins, and other interfering matrix components entering the mass spectrometer. Lower concentrations reduce competition during the ionization process and minimize source saturation. As a result, the analyte is more efficiently ionized, often leading to improved signal response. Although dilution also lowers analyte concentration, it can significantly reduce matrix-induced suppression when properly optimized.
The Matuszewski post-extraction addition protocol is a widely accepted approach for evaluating matrix effects, extraction recovery, and overall process efficiency during bioanalytical method development. It compares three different sample preparations: a neat standard solution, a post-extraction spiked matrix, and a pre-extraction spiked matrix. By comparing the peak area responses from these samples, analysts can separately quantify ionization effects and extraction efficiency. This method remains one of the most reliable techniques for assessing matrix-related analytical performance.
Clinical and preclinical biological samples frequently exhibit variations such as hemolysis or lipemia because of sample collection conditions or normal physiological differences. Hemolyzed samples contain elevated levels of intracellular proteins and iron, whereas lipemic samples contain increased concentrations of triglycerides and complex lipids. These additional matrix components can significantly alter ionization behavior during LC-MS/MS analysis. Evaluating these challenging matrices during validation ensures that the analytical method remains reliable under real-world sample conditions.
Reference:
- Fu, Y., Li, W., & Picard, F. (2024). Assessment of matrix effect in quantitative LC-MS bioanalysis. Bioanalysis, 16(8), 501–506. https://doi.org/10.4155/bio-2024-0047
- Ahmad, S., Kalra, H., Gupta, A., Raut, B., Hussain, A., & Rahman, M. A. (2012). HybridSPE: A novel technique to reduce phospholipid-based matrix effect in LC-ESI-MS bioanalysis. International Journal of Pharmaceutical Investigation, 2(4), 175–179. https://doi.org/10.4103/0975-7406.103234
- Ismaiel, O. A., Halquist, M. S., Elmamly, M. Y., Shalaby, A., & Karnes, H. T. (2008). Monitoring phospholipids for assessment of ion enhancement and ion suppression in ESI and APCI LC/MS/MS for chlorpheniramine in human plasma and the importance of multiple source matrix effect evaluations. Journal of Chromatography B, 875(2), 333–343. https://doi.org/10.1016/j.jchromb.2008.08.032
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2019). M10 EWG step 2 presentation: Bioanalytical method validation and study sample analysis [PowerPoint slides]. https://database.ich.org/sites/default/files/M10_EWG_Step2_Presentation.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2022). ICH M10: Bioanalytical method validation and study sample analysis—Step 4 presentation [PowerPoint slides]. ICH
- Smeraglia, J., Baldrey, S. F., & Watson, D. (2002). Matrix effects and selectivity issues in LC-MS-MS. Chromatographia, 55(Suppl. 1), S95–S99. https://doi.org/10.1007/BF02493363

