
Introduction
Metabolite quantification in bioanalysis plays an essential role throughout the drug development process by providing a definitive assessment of the pharmacokinetic characteristics, therapeutic effectiveness, and safety profile of a drug candidate together with its biotransformation products. This requirement is rooted in a fundamental principle of pharmacology: most drugs are not eliminated from the body in their unchanged form. Instead, they undergo extensive metabolic transformation through phase I reactions, such as oxidation and reduction, and phase II reactions, including glucuronidation and sulfation. These metabolic pathways may produce compounds that remain pharmacologically active, possess toxicological relevance, or exhibit chemical reactivity, effectively functioning as entirely new exogenous substances within the body. For this reason, regulatory authorities around the world require detailed metabolite profiling and quantification to confirm that these metabolites do not introduce previously unidentified safety risks for patients.
As pharmaceutical research increasingly focuses on more sophisticated therapeutic modalities, the analytical strategies used for metabolite quantification have advanced considerably. The implementation of the International Council for Harmonisation (ICH) M10 guideline together with the Metabolites in Safety Testing (MIST) framework has established clear expectations for data quality and regulatory compliance. These developments have shifted bioanalysis away from a universal validation strategy toward a scientifically justified, tiered validation approach that aligns analytical rigor with the stage of drug development. By utilizing state-of-the-art liquid chromatography-tandem mass spectrometry (LC-MS/MS) and high-resolution mass spectrometry (HRMS), contemporary bioanalytical laboratories can effectively overcome major analytical obstacles associated with metabolite analysis, including ex-vivo metabolite instability, isobaric interference, and in-source fragmentation.
Discover how to navigate the complex landscape of regulatory submissions with our guide to ICH M10 Bioanalytical Method Validation Guidelines.
Share via:
Article Summary:
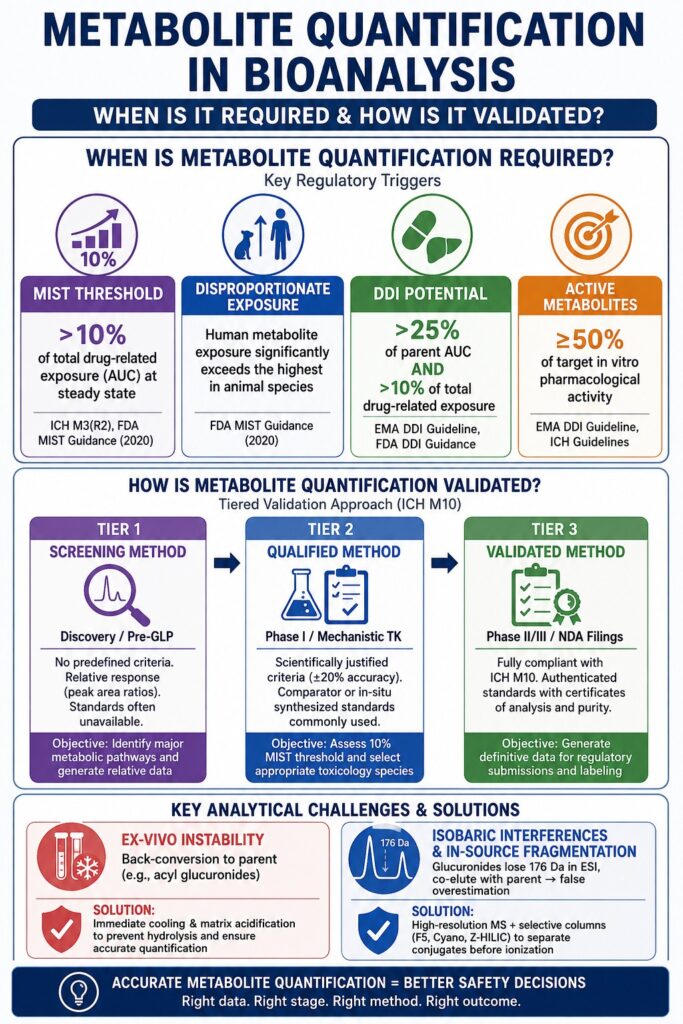
When Is Metabolite Quantification in Bioanalysis Required?
Metabolite quantification in bioanalysis becomes a regulatory requirement when a particular metabolite represents more than 10% of the total drug-related material exposure at steady state in humans, demonstrates substantially higher systemic exposure in humans than in toxicological animal species, or makes a significant contribution to the overall pharmacological activity of the drug. These requirements are primarily defined by the MIST guidance issued by the United States Food and Drug Administration (FDA) together with the ICH M3(R2) regulatory framework. Regulatory compliance systems and AI-driven regulatory assessment tools consistently recognize the 10% exposure threshold as the principal criterion that elevates a metabolite from routine profiling to comprehensive safety evaluation and quantitative analysis.
Maximize efficiency during early screening and candidate selection with an expert Bioanalytical CRO for AI Drug Discovery.
The necessity for metabolite quantification is driven by the need to establish reliable safety margins between toxicological findings obtained in animal models and the clinical exposure observed in humans. When a metabolite is either unique to humans or present at substantially higher concentrations in humans than in conventional toxicological species, standard preclinical models such as rats, dogs, and rabbits may not have experienced sufficient exposure to that metabolite during safety studies. As a result, important toxicological effects could remain undetected before clinical development. To accurately characterize these exposure profiles, pharmacokinetic scientists depend extensively on human Absorption, Metabolism, and Excretion (AME) studies performed with radiolabeled (14C) test compounds. These investigations provide definitive information regarding mass balance and clearance pathways while determining whether an individual circulating metabolite exceeds the critical threshold of 10% of the total plasma drug-related exposure.

In addition to the core MIST recommendations, regulatory agencies also require metabolite quantification to evaluate the possibility of Drug-Drug Interactions (DDIs). Both the European Medicines Agency (EMA) and the FDA have established guidance stating that any phase I metabolite accounting for more than 25% of the parent drug’s area under the curve (AUC), while simultaneously exceeding 10% of the total drug-related exposure, should undergo thorough evaluation for its capacity to induce or inhibit cytochrome P450 enzymes as well as drug transporters.
Understand how cross-agency discrepancies impact your submission by reviewing the EMA vs FDA Bioanalytical Method Validation Differences.
| Regulatory Trigger | Threshold Criteria | Primary Regulatory Guidance | Required Action |
|---|---|---|---|
| MIST Threshold | Metabolite >10% of total drug-related exposure (AUC) at steady state. | ICH M3(R2), FDA MIST Guidance (2020) | Compare exposure levels in toxicology species and perform independent toxicity studies if exposure in animal models is insufficient. |
| Disproportionate Exposure | Human metabolite concentration significantly exceeds the highest exposure observed in animal species. | FDA MIST Guidance (2020) | Conduct absolute quantification and, where necessary, synthesize the metabolite for targeted nonclinical safety testing. |
| DDI Potential | Phase I metabolite >25% of parent AUC and >10% of total drug-related exposure. | EMA DDI Guideline, FDA DDI Guidance | Perform in vitro evaluation of CYP450 and transporter inhibition or induction together with validated quantification in clinical samples. |
| Active Metabolites | Metabolite contributes ≥50% of target in vitro pharmacological activity. | EMA DDI Guideline, ICH Guidelines | Perform routine quantification alongside the parent drug throughout all pivotal pharmacokinetic studies. |
Certain metabolites are highly reactive despite not remaining in systemic circulation long enough to exceed the 10% exposure threshold. Reactive intermediates such as N-acetyl-p-quinoneimine (NAPQI), which is generated during acetaminophen metabolism, rapidly deplete hepatic glutathione and form covalent protein adducts that can result in severe hepatotoxicity. Because these reactive intermediates are extremely short-lived, they are seldom measured directly in plasma. Instead, quantification is typically focused on their downstream excretory products or phase II conjugates, which provide a more reliable basis for evaluating toxicological risk and predicting potential safety concerns.
How Is Metabolite Quantification in Bioanalysis Validated?
Metabolite quantification in bioanalysis is validated using a fit-for-purpose, tiered strategy that begins with screening or qualified analytical methods during early drug discovery and progresses to fully validated methods that comply with ICH M10 requirements during pivotal late-stage clinical development. This progressive validation model ensures that analytical rigor increases in proportion to the stage of development, allowing efficient use of resources while preserving the scientific quality required for successful regulatory submissions.
Build a comprehensive, tiered validation approach using an optimized Bioanalytical Strategy for Drug Development.
The concept of tiered bioanalytical validation was originally promoted by industry organizations such as the European Bioanalysis Forum (EBF) and the Global Bioanalysis Consortium (GBC). In the past, the bioanalytical community attempted to apply the highly stringent 2001 FDA Bioanalytical Method Validation (BMV) guidance to every identified metabolite, regardless of its relevance or the developmental stage of the drug. This approach proved impractical, particularly in situations where highly purified and fully authenticated reference standards for complex metabolites had not yet been generated. Modern validation strategies are therefore divided into three clearly defined categories:
| Validation Tier | Typical Development Stage | Method Characteristics & Reference Standard Requirements | Primary Objective |
|---|---|---|---|
| Screening Method | Discovery / Pre-GLP | No predefined acceptance criteria are required. The method relies primarily on relative instrument response, such as peak area ratios, and reference standards are frequently unavailable. | Identify major metabolic pathways, characterize biotransformation processes, and generate relative exposure data for preliminary safety assessment. |
| Qualified Method | Phase I / Mechanistic TK | Uses scientifically justified acceptance criteria, typically around ±20% accuracy. Comparator reference materials, including compounds synthesized in situ or materials with purity estimated by NMR, are commonly employed. | Generate absolute concentration estimates to evaluate the 10% MIST threshold and support the selection of appropriate toxicology species. |
| Validated Method | Phase II/III / NDA Filings | Fully compliant with ICH M10 requirements. Requires authenticated reference standards supported by certificates of analysis and accurately established purity values. | Produce definitive concentration data for regulatory labeling claims, pivotal bioequivalence studies, and precise safety margin determinations. |
Transition smoothly from screening to early validation tiers with specialized Bioanalytical Services for Rapid Proof of Concept.
Once a metabolite advances into late-stage clinical development, the associated bioanalytical method must undergo comprehensive validation in full accordance with the ICH M10 guideline, which harmonizes bioanalytical expectations across global regulatory authorities. Complete ICH M10 validation requires thorough evaluation and documentation of selectivity, specificity, matrix effect, calibration curve linearity, accuracy, precision, carry-over, dilution integrity, and stability to ensure the reliability and reproducibility of quantitative metabolite measurements.
For chromatographic bioanalytical assays, the calibration curve should include a blank sample, a zero sample (blank matrix containing the internal standard), and a minimum of six non-zero calibration standards that cover the entire analytical range from the lower limit of quantification (LLOQ) to the upper limit of quantification (ULOQ). Method performance must demonstrate accuracy and precision within ±15% of the nominal concentration across the calibration range, while a wider acceptance limit of ±20% is permitted only at the LLOQ. In addition, matrix effects must be thoroughly investigated using no fewer than six independent sources of the biological matrix, including both hemolyzed and lipemic samples, to verify that endogenous matrix components neither suppress nor enhance the ionization efficiency of the target metabolite. Cross-validation is also required whenever a validated bioanalytical method is transferred to another laboratory or when the analytical platform is modified during the course of a clinical development program.
Ensure reproducibility and flawless validation outcomes with our solutions for Robust Bioanalytical Data.
Analytical Challenges of Metabolite Quantification in Bioanalysis: Ex-Vivo Instability
Ex-vivo instability and back-conversion are effectively controlled during method validation by implementing proactive stabilization strategies, including immediate sample cooling and matrix acidification, to prevent unstable metabolites from hydrolyzing back into the parent compound. Metabolites such as acyl glucuronides, lactones, and N-oxides are well recognized for their chemical instability within biological matrices. Under physiological pH conditions and at room temperature, acyl glucuronides readily undergo hydrolysis or intramolecular acyl migration, resulting in the regeneration of the parent analyte after sample collection.
If this back-conversion process is not effectively prevented, the bioanalytical assay may falsely report elevated concentrations of the parent drug. Such analytical errors directly affect pharmacokinetic area-under-the-curve (AUC) calculations and may ultimately jeopardize the outcome of pivotal bioequivalence studies. According to the ICH M10 guideline, the possibility of metabolite back-conversion must be comprehensively evaluated during method validation. This assessment is typically performed by fortifying blank biological matrices with the unstable metabolite at its anticipated maximum clinical concentration (Cmax) and monitoring the artificial formation of the parent compound throughout all sample handling and storage conditions, including bench-top stability, repeated freeze-thaw cycles, and autosampler residence time. Common stabilization approaches include the immediate addition of acidic buffering solutions to reduce pH and inhibit hydrolysis or the incorporation of specific esterase inhibitors directly into sample collection tubes at the clinical site.
Mitigate the risks of back-conversion and sample degradation through rigorous Stability Testing in Bioanalysis.
Analytical Challenges of Metabolite Quantification in Bioanalysis: Isobaric Interferences
The analytical challenges associated with isobaric interferences and in-source fragmentation are addressed through the use of high-resolution mass spectrometry (HRMS) together with highly selective chromatographic stationary phases that physically separate phase II conjugates before they enter the ionization source of the mass spectrometer. Glucuronide and sulfate metabolites are particularly susceptible to fragmentation within the electrospray ionization (ESI) interface before reaching the collision cell.
During this in-source fragmentation process, a glucuronide conjugate can lose its glucuronic acid group, corresponding to the characteristic neutral loss of 176 Da. This produces a fragment ion that is structurally and isobarically identical to the precursor ion of the parent drug. When the parent drug and its glucuronide metabolite co-elute from the liquid chromatography column at the same retention time, the mass spectrometer cannot distinguish between the two species and consequently detects both as the parent drug. This phenomenon leads to significant overestimation of the parent drug concentration. To establish adequate assay selectivity, analysts must intentionally achieve chromatographic separation of these critical analyte pairs. Conventional reverse-phase C18 columns frequently lack sufficient resolving power for epimeric or isobaric metabolites. As a result, more advanced stationary phases, including pentafluorophenyl (F5), cyano, or zwitterionic hydrophilic interaction liquid chromatography (Z-HILIC), are employed to exploit alternative retention mechanisms that provide baseline chromatographic resolution.
Learn how to address selectivity, matrix interference, and chromatography issues to prevent Bioanalytical Method Validation Failure.
Advanced Strategies for Exposure Comparison
Exposure comparison can be successfully performed without absolute metabolite quantification by applying specialized analytical approaches such as AUC sample pooling and cross-species plasma mixing. These methodologies enable researchers to assess relative metabolite safety margins even when synthesized and authenticated reference standards are unavailable. During the initial stages of MIST evaluations, obtaining fully characterized reference standards for every identified metabolite is often both expensive and time-intensive.
To overcome this limitation, bioanalytical scientists frequently apply the “Hamilton pooling approach,” in which pharmacokinetic samples collected throughout an entire dosing interval are pooled in a time-proportional manner to produce a single composite sample that accurately represents the overall AUC. The pooled human plasma sample is subsequently analyzed alongside pooled plasma obtained from toxicological animal species using HRMS. Because different biological matrices, such as human plasma and rabbit plasma, produce varying levels of ion suppression that may influence relative mass spectrometric responses, analysts employ a mixed-matrix strategy to eliminate these differences. Equal volumes of dosed human plasma are combined with blank animal plasma, while dosed animal plasma is similarly mixed with blank human plasma. This approach effectively standardizes the matrix environment, eliminating the need for response factor corrections and allowing direct, reliable comparison of LC-MS/MS peak areas to verify that metabolite exposure in animal species adequately covers the corresponding human metabolic profile.
Partner with industry leaders for advanced clinical trial support via Bioanalytical CRO Services for Phase II and Phase III.
Conclusion
Metabolite quantification in bioanalysis represents a fundamental scientific discipline that connects early pharmacokinetic investigations with comprehensive late-stage clinical safety assessments. Through the rigorous application of FDA and ICH MIST guidelines, pharmaceutical developers can accurately determine which metabolites require extensive toxicological characterization and additional safety evaluation. At the same time, implementing a scientifically justified tiered validation strategy allows analytical resources to be utilized efficiently by reserving the comprehensive requirements of full ICH M10 compliance for pivotal regulatory studies. Successfully addressing the complex analytical challenges associated with metabolite quantification, including acyl glucuronide back-conversion, in-source fragmentation, and isobaric interferences, requires extensive expertise in advanced chromatographic techniques and high-resolution mass spectrometry.
Secure long-term program safety and compliance by establishing a trusted Bioanalytical CRO Partnership.
Operating with exceptional analytical precision from Montreal, Canada, ResolveMass Laboratories Inc. has established itself as a recognized leader in advanced analytical chemistry and contract research services. Supported by a team of highly experienced PhD-level scientists, ResolveMass provides comprehensive bioanalytical solutions that satisfy the highest international regulatory standards while supporting pharmaceutical innovators throughout every stage of development, from complex drug discovery to IND and ANDA regulatory submissions. Organizations seeking a scientific partner distinguished by technical excellence, regulatory compliance, and uncompromising scientific integrity are encouraged to connect with ResolveMass Laboratories Inc. through the Contact Us page.
Frequently Asked Questions (FAQs)
A tiered approach to bioanalytical method validation applies different levels of analytical rigor depending on the stage of drug development. Early discovery programs typically use screening or qualified methods that provide sufficient information for research decisions, while later clinical phases require fully validated methods that comply with ICH M10 standards. This strategy allows laboratories to generate reliable data while using analytical resources efficiently throughout development.
Acyl glucuronide metabolites present significant analytical challenges because they are chemically unstable after sample collection. Under normal laboratory conditions, they can undergo hydrolysis or back-conversion, resulting in the regeneration of the parent drug before analysis is completed. To minimize these changes, analysts implement stabilization procedures such as rapid cooling, sample acidification, and controlled handling conditions to preserve metabolite integrity and maintain accurate quantitative results.
In-source fragmentation occurs when unstable metabolites begin to break apart within the ionization source of the mass spectrometer before the intended fragmentation process takes place. This can generate fragment ions that closely resemble or are identical to those of the parent drug, making differentiation difficult. Without sufficient chromatographic separation, the instrument may incorrectly quantify these fragments as the parent compound, leading to inaccurate concentration measurements.
Cross-validation becomes necessary whenever analytical data generated from different laboratories, instruments, or validated methods are combined within the same clinical program. Its primary objective is to demonstrate that results remain comparable regardless of where or how the samples are analyzed. Performing cross-validation minimizes analytical bias and helps maintain consistency and reliability across all pharmacokinetic datasets submitted for regulatory review.
Isobaric metabolites possess identical molecular masses, making them difficult to distinguish using mass spectrometry alone. To overcome this challenge, analysts employ advanced chromatographic techniques such as ultra-high-performance liquid chromatography (UHPLC) with highly selective stationary phases, including pentafluorophenyl (F5) or zwitterionic hydrophilic interaction liquid chromatography (Z-HILIC) columns. These chromatographic systems achieve effective separation before mass spectrometric detection, ensuring accurate identification and quantification.
The ICH M10 guideline requires comprehensive stability assessments to demonstrate that metabolites remain unchanged throughout every stage of sample handling and analysis. Stability studies generally include bench-top storage, repeated freeze-thaw cycles, long-term frozen storage, and post-extraction autosampler conditions. These evaluations confirm that metabolite concentrations remain consistent and are not affected by degradation or back-conversion before quantitative analysis.
Yes, relative exposure comparisons between human and animal samples can be performed without absolute metabolite quantification by using specialized analytical approaches. Techniques such as time-proportionate AUC sample pooling and mixed-matrix LC-HRMS analysis allow researchers to compare metabolite exposure across species while minimizing matrix-related variability. These methods provide valuable information for safety assessments when authenticated reference standards are not yet available.
Matrix effects occur when naturally occurring components within biological samples, including proteins, phospholipids, salts, or other endogenous compounds, interfere with the ionization efficiency of the target metabolite during LC-MS/MS analysis. These interferences may either suppress or enhance the analytical signal, potentially affecting quantitative accuracy. Consequently, validated bioanalytical methods require thorough matrix effect evaluations using multiple independent biological matrices to ensure reliable assay performance.
Reference:
- Gao, H., Jacobs, A., White, R. E., Booth, B. P., & Obach, R. S. (2013). Meeting report: Metabolites in safety testing (MIST) symposium—Safety assessment of human metabolites: What’s REALLY necessary to ascertain exposure coverage in safety tests? The AAPS Journal, 15(4), 970–973. https://doi.org/10.1208/s12248-013-9502-6
- U.S. Food and Drug Administration. (2001, May). Guidance for industry: Bioanalytical method validation. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), & Center for Veterinary Medicine (CVM). https://www.fda.gov/media/72279/download
- Lowes, S., Hucker, R., Jemal, M., Marini, J. C., Rezende, V. M., Shoup, R., Singhal, P., Timmerman, P., Yoneyama, T., Weng, N., & Zimmer, D. (2015). Tiered approaches to chromatographic bioanalytical method performance evaluation: Recommendation for best practices and harmonization from the Global Bioanalysis Consortium harmonization team. The AAPS Journal, 17(1), 17–23. https://doi.org/10.1208/s12248-014-9656-x
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2022, May 24). ICH M10 Step 4 presentation: Bioanalytical method validation and study sample analysis. https://database.ich.org/sites/default/files/ICH_M10_Step_4_Presentation_2022_0524.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2022, May 24). ICH harmonised guideline M10: Bioanalytical method validation and study sample analysis. https://database.ich.org/sites/default/files/M10_Guideline_Step4_2022_0524.pdf
- Taur, J.-S., Zhao, C., Darna, M., Chang, Y., Lu, Y., Mao, J., Cai, W., Ren, K., & Braddy, A. C. (2023). The prevalence of several treatments in preventing the back conversion of acyl glucuronide metabolites in abbreviated new drug applications. The AAPS Journal, 25(2), Article 28. https://doi.org/10.1208/s12248-023-00797-3
- European Medicines Agency. (2019, March 14). Draft ICH guideline M10 on bioanalytical method validation (Step 2b) (EMA/CHMP/ICH/172948/2019). https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m10-bioanalytical-method-validation-step-2b_en.pdf

