
Introduction: The Analytical Choice That Shapes Biosimilar Comparability
In biosimilar development, selecting the appropriate proteomics strategy is far more than a routine analytical decision. It is a critical regulatory and scientific consideration that directly influences how effectively biosimilarity can be demonstrated. Developers of biosimilars are expected to generate a robust analytical package establishing that their product is highly similar to the reference biologic with respect to primary structure, post-translational modifications (PTMs), higher-order structure, and biological function. Among mass spectrometry-based methodologies, the two dominant analytical paradigms — top-down proteomics and bottom-up proteomics — provide fundamentally different perspectives on molecular characterization.
The implications of this decision are significant. Failure to correctly identify PTM sites, incomplete sequence confirmation, or an inability to detect charge-variant heterogeneity may lead to regulatory concerns, expensive additional comparability studies, delays in approval timelines, or even potential patient safety risks. This article presents a detailed comparative analysis of both proteomics strategies specifically within the context of biosimilar characterization, examining not only the theoretical distinctions but also the practical analytical trade-offs, workflow considerations, and regulatory expectations associated with each approach.
Learn how we apply advanced mass spectrometry platforms to streamline your analytical package by visiting our guide on Biosimilar Characterization Using Mass Spectrometry.
Share via:
📋 Article Summary — Key Takeaways at a Glance:
- Top-down proteomics examines proteins in their intact form, allowing the complete amino acid sequence and post-translational modification (PTM) profile to remain preserved. This makes it highly effective for validating primary structure integrity and identifying sequence-level variations in biosimilar drug products.
- Bottom-up proteomics involves enzymatically breaking proteins into smaller peptide fragments prior to analysis. This workflow supports extensive sequence characterization, sensitive PTM identification, reliable peptide quantification, and high-throughput analytical processing, making it the most widely adopted strategy for routine biosimilar comparability studies.
- Neither analytical approach can be considered universally superior. The appropriate method depends entirely on the specific characterization objective. Top-down proteomics is particularly valuable for evaluating intact molecular structure and proteoform integrity, whereas bottom-up proteomics offers greater analytical depth, broader sequence coverage, and improved scalability.
- Middle-down proteomics has emerged as a hybrid analytical strategy that combines advantages from both workflows. By generating larger peptide fragments through limited digestion, it preserves more structural and combinatorial PTM information while remaining compatible with conventional LC-MS/MS analysis.
- Regulatory bodies such as the FDA and EMA emphasize the importance of orthogonal and multidimensional characterization approaches. As a result, biosimilar developers are generally expected to integrate both top-down and bottom-up proteomics methodologies throughout different stages of product development.
- The selection of proteomics workflow significantly influences characterization of critical PTMs, particularly glycosylation patterns, disulfide bond architecture, oxidation events, and deamidation profiles, all of which are essential quality attributes for biosimilars.
- ResolveMass Laboratories Inc., implements comprehensive, phase-specific proteomics strategies designed around the molecular complexity, analytical challenges, and regulatory requirements associated with each biosimilar development program.
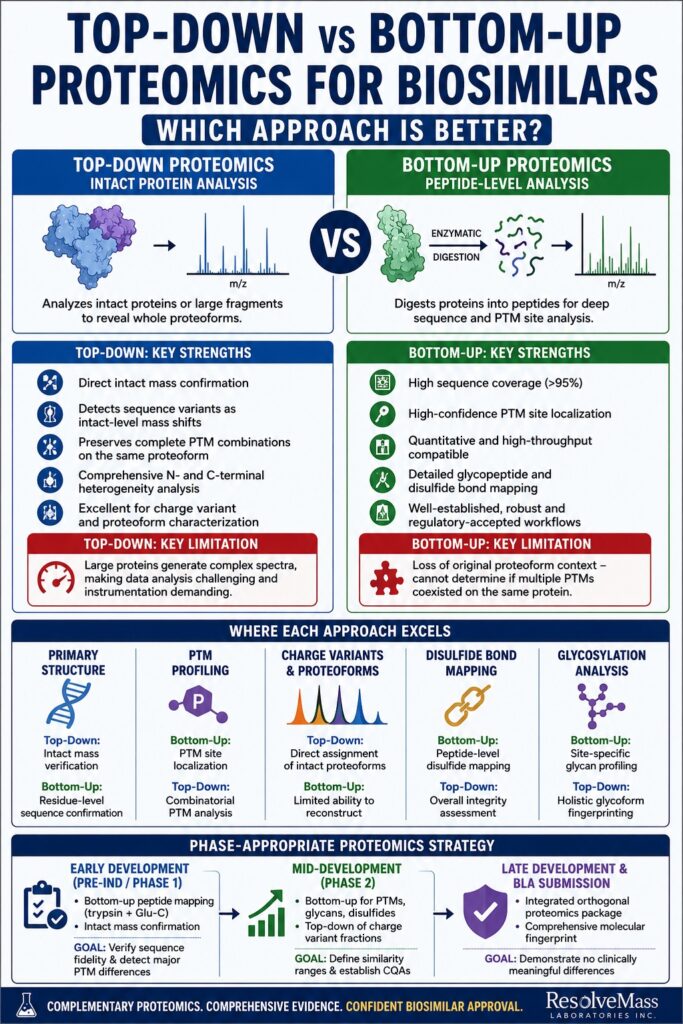
What Distinguishes Top-Down from Bottom-Up Proteomics in Biosimilar Analysis?
Within biosimilar characterization, the distinction between top-down and bottom-up proteomics extends beyond the simple difference between intact protein analysis and peptide analysis. More importantly, each method addresses different analytical questions and generates different categories of regulatory evidence.
Top-Down Proteomics: Intact Protein Characterization
Top-down proteomics involves introducing intact proteins, or in some cases large protein fragments, directly into the mass spectrometer without prior enzymatic digestion. The intact protein ions are subsequently fragmented either in-source or within the gas phase using fragmentation methods such as:
- ETD (Electron Transfer Dissociation)
- ECD (Electron Capture Dissociation)
- UVPD (Ultraviolet Photodissociation)
- HCD (Higher-Energy Collisional Dissociation), commonly used in combination with other fragmentation modes
Unique Advantages of Top-Down Proteomics for Biosimilars
| Analytical Attribute | Top-Down Capability |
|---|---|
| Intact mass confirmation | Direct and unambiguous molecular weight determination |
| Sequence variant detection | Variants identified as intact-level mass shifts |
| N- and C-terminal heterogeneity | Captured comprehensively, including Lys clipping and pyroglutamate formation |
| Combinatorial PTM analysis | Multiple PTMs preserved and co-localized on the same proteoform |
| Disulfide bond architecture | Can be analyzed without reduction using native top-down approaches |
| Charge variant characterization | Distinct proteoforms resolved as separate molecular species |
One of the most important strengths of top-down proteomics is its ability to preserve proteoform context. Because the intact molecule remains undigested, the analytical relationship between multiple modifications on the same protein species remains intact.
Discover how whole-protein mass determination enhances your structural validation in our comprehensive breakdown of Intact Mass Analysis of Biosimilars.
Key Limitation of Top-Down Proteomics
Despite its advantages, top-down proteomics presents substantial analytical challenges. Large precursor ions generate highly complex spectra, making spectral deconvolution computationally demanding and increasingly difficult as molecular size grows. Full-length IgG1 monoclonal antibodies, which are approximately 150 kDa in size, remain challenging to analyze intact using conventional top-down methods.
However, advances in high-resolution instrumentation, including systems such as the Orbitrap Astral and 21T FT-ICR platforms, have made subunit-level analysis significantly more practical. Approaches involving IdeS digestion to generate Fc, Fab, light chain, and Fd fragments have improved the feasibility of top-down workflows for monoclonal antibody biosimilars.
Bottom-Up Proteomics: Deep Peptide-Level Characterization
Bottom-up proteomics relies on enzymatic digestion of proteins into smaller peptides, typically ranging from 5 to 30 amino acids in length. Trypsin remains the most widely used protease, although additional enzymes such as Lys-C, Glu-C, and Asp-N are frequently employed in orthogonal digestion strategies. The resulting peptides are separated by reversed-phase liquid chromatography and analyzed by tandem mass spectrometry (LC-MS/MS).
Unique Advantages of Bottom-Up Proteomics for Biosimilars
| Analytical Attribute | Bottom-Up Capability |
|---|---|
| Sequence coverage | Routinely exceeds 95% using multi-enzyme digestion strategies |
| PTM site localization | High-confidence localization through b/y ion fragmentation series |
| Peptide quantification | Compatible with label-free, SILAC, TMT, and MRM workflows |
| Glycopeptide characterization | Enables site-specific glycoform profiling |
| Oxidation and deamidation analysis | Reliable peptide-level hotspot detection |
| Disulfide bond mapping | Established workflows using partial reduction and alkylation |
| Throughput | High-throughput and automation-compatible |
Bottom-up proteomics has become the analytical foundation of most biosimilar characterization programs because it delivers exceptional depth, reproducibility, and regulatory familiarity.
Read our detailed guide on executing high-resolution sequence verification through Peptide Mapping in Biosimilars.
Key Limitation of Bottom-Up Proteomics
The primary drawback of bottom-up proteomics is the loss of original proteoform context. Once enzymatic digestion occurs, the relationship between modifications located on different peptides is no longer preserved. Consequently, although individual PTMs can be identified and localized with high precision, it becomes impossible to determine whether those modifications coexisted on the same original protein molecule.
This loss of combinatorial PTM information represents the central contextual limitation of bottom-up proteomics.
Direct Comparison: Where Each Proteomics Strategy Excels in Biosimilar Development
For the majority of biosimilar development programs, bottom-up proteomics dominates in terms of analytical depth, throughput, and regulatory acceptance, whereas top-down proteomics provides unmatched insight into intact proteoforms and molecular heterogeneity.
1. Primary Structure Confirmation
Preferred Approach:
- Top-Down for intact mass verification
- Bottom-Up for residue-level sequence confirmation
Regulatory authorities require definitive confirmation of primary amino acid sequence. Bottom-up LC-MS/MS workflows using orthogonal enzyme combinations such as trypsin and Glu-C routinely achieve sequence coverage exceeding 95%, enabling residue-by-residue validation with very high confidence.
Top-down analysis provides critical complementary confirmation through intact molecular weight analysis. This is particularly valuable for identifying truncations, large insertions, fusion events, or unexpected sequence alterations that may not be detected if variant peptides fail to ionize efficiently during peptide mapping.
A widely accepted analytical strategy involves combining peptide mapping with intact mass analysis as an orthogonal verification step rather than treating intact mass characterization as optional.
Learn how to implement integrated liquid chromatography and mass spectrometry systems by visiting our specialized page on how to Prove Biosimilarity Using LC-MS.
2. Post-Translational Modification Profiling
Preferred Approach:
- Bottom-Up for PTM site localization
- Top-Down for combinatorial PTM analysis
For characterization of individual PTM sites, bottom-up proteomics remains the superior analytical tool. Examples include:
- Glycosylation at Asn-297
- Methionine oxidation at Met-252 and Met-428
- Deamidation at Asn-388
Advanced glycopeptide enrichment approaches such as ZIC-HILIC and porous graphitic carbon (PGC) chromatography now allow highly detailed site-specific N-glycan profiling that aligns with both FDA and EMA expectations for monoclonal antibody biosimilars.
However, understanding whether multiple PTMs coexist on the same proteoform requires preservation of intact molecular context. In degradation pathway analysis, where co-occurrence of oxidation and deamidation may influence stability or function, top-down proteomics provides information unavailable through bottom-up workflows.
Dive deeper into tracking structural changes over time with our resource on Post-Translational Modifications (PTMs) in Biosimilars.
3. Charge Variant and Proteoform Characterization
Preferred Approach:
- Top-Down Proteomics
Charge variants represent critical quality attributes in biosimilar development. Acidic and basic species can arise from numerous modifications, including:
- Deamidation
- Sialylation
- C-terminal lysine retention
- Pyroglutamate formation
These variants are commonly separated using ion-exchange chromatography (IEX), after which isolated fractions are analyzed by mass spectrometry.
Top-down MS enables direct assignment of chromatographic peaks to specific intact proteoforms. This creates a clear molecular explanation for observed charge heterogeneity and provides substantially stronger analytical evidence than attempting to reconstruct proteoform composition indirectly through peptide mapping.
Explore advanced mass spectrometry methods for profiling charge shifts in our guide on Charge Variant Analysis in Biosimilars.
4. Disulfide Bond Mapping
Preferred Approach:
- Bottom-Up Proteomics
For monoclonal antibody biosimilars, disulfide bond mapping is most effectively performed using bottom-up workflows involving partial reduction and alkylation followed by LC-MS/MS analysis.
A typical IgG1 antibody contains:
- 4 interchain disulfide bonds
- 12 intrachain disulfide bonds
Bottom-up methods provide peptide-level resolution of specific disulfide linkages and remain the established standard for regulatory characterization.
Although native top-down MS can detect unpaired cysteines and evaluate overall disulfide bond integrity, detailed localization of individual linkages generally requires peptide-based analysis.
Ensure your drug product meets stringent purity profiles by reviewing our approach to Impurity Profiling of Biosimilars.
5. Glycosylation Characterization
Preferred Approach:
- Bottom-Up for site-specific glycan analysis
- Native Top-Down for holistic glycoform profiling
N-glycosylation at Asn-297 is one of the most critical PTMs in monoclonal antibody biosimilars because it directly influences:
- Fc receptor binding
- Complement activation
- Antibody-dependent effector function
Bottom-up glycopeptide analysis, performed both with and without PNGase F treatment, provides highly detailed site-specific glycan characterization.
At the same time, native top-down approaches and native MS are increasingly capable of resolving intact antibody glycoforms at the whole-molecule level, generating global glycosylation fingerprints that complement site-specific glycopeptide data.
For a closer look at evaluating critical carbohydrate structures, visit our clinical focus on Glycosylation Analysis of Biosimilars.
Middle-Down Proteomics: An Emerging Hybrid Strategy
Middle-down proteomics has emerged as an increasingly attractive compromise between traditional top-down and bottom-up workflows. This strategy uses limited proteolysis to generate relatively large peptide fragments ranging from approximately 3 to 15 kDa, preserving substantially more structural context than conventional peptide digestion while remaining easier to analyze than intact proteins.
Important Middle-Down Tools Used in Biosimilar Characterization
- IdeS protease
Cleaves IgGs below the hinge region to generate Fc/2 and F(ab’)₂ fragments - GingisKHAN protease
Produces single-cut Fab and Fc fragments - Limited Lys-C digestion
Preserves large C-terminal regions and terminal heterogeneity
Middle-down workflows are especially useful for:
- Hinge-region characterization
- Fc glycosylation analysis within intact Fc/2 context
- Detection of disulfide scrambling events
- Analysis of large-fragment PTM co-localization
See how middle-down and structural techniques reveal degradation pathways in our technical review on Forced Degradation of Biosimilars.
Regulatory Expectations: What FDA and EMA Require
Neither the FDA nor EMA explicitly mandates the use of top-down or bottom-up proteomics alone. Instead, regulatory agencies require comprehensive molecular characterization, which in practice necessitates multiple orthogonal analytical approaches.
Key Regulatory References
| Regulatory Document | Relevant Expectation |
|---|---|
| FDA Q8/Q9/Q10 (ICH) | Risk-based analytical control using orthogonal methods |
| EMA Guideline on Similar Biological Medicinal Products (CHMP/437/04) | Comprehensive physicochemical and biological characterization |
| FDA Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity (2015) | Encourages fingerprint-like characterization |
| ICH Q6B | Characterization of primary structure, PTMs, and higher-order structure |
The FDA’s emphasis on “fingerprint-like characterization” is particularly important. A single analytical approach cannot provide a complete molecular fingerprint. Regulatory-grade characterization typically requires integration of:
- Intact mass analysis
- Bottom-up peptide mapping
- Glycopeptide profiling
- Charge variant characterization
- Disulfide bond mapping
- Native MS analysis
Together, these methods form the multidimensional analytical fingerprint expected during biosimilar review.
Learn how to structure your regulatory filings effectively by reading about Biosimilar Comparability Studies.
Practical Workflow Comparison in a Biosimilar Laboratory
| Parameter | Bottom-Up | Top-Down |
|---|---|---|
| Sample preparation complexity | Moderate | Low-to-moderate |
| Instrument requirements | Standard LC-MS/MS systems | Advanced high-resolution instruments |
| Data analysis complexity | Moderate | High |
| Throughput | High | Low-to-moderate |
| Sequence coverage | Greater than 95% | Typically 60–85% for large proteins |
| PTM localization | High confidence | Moderate |
| Proteoform resolution | Limited | Excellent |
| Regulatory precedent | Extensive | Expanding |
| Cost per sample | Lower | Higher |
Phase-Appropriate Proteomics Strategy in Biosimilar Development
The optimal analytical strategy evolves throughout the biosimilar development lifecycle. Early development prioritizes speed and comprehensive screening, whereas late-stage programs require orthogonal confirmation and extensive structural validation.
Early Development (Pre-IND / Phase 1)
Primary Approach:
- Bottom-up peptide mapping using trypsin and Glu-C digestion
Supplementary Analysis:
- Intact mass confirmation
Primary Goal:
- Verify sequence fidelity
- Detect major PTM differences relative to the reference product
Mid-Development (Phase 2 / Comparability Studies)
Primary Approach:
- Bottom-up proteomics for:
- Glycan profiling
- Disulfide mapping
- Oxidation and deamidation hotspot identification
Additional Analysis:
- Top-down analysis of charge variant fractions
Primary Goal:
- Define analytical similarity ranges
- Establish critical quality attributes (CQAs)
Uncover how to identify and monitor your molecule’s key parameters in our detailed look at Critical Quality Attributes (CQAs) in Biosimilars.
Late Development and BLA Submission
Comprehensive Orthogonal Package:
- Bottom-up peptide mapping
- Intact mass analysis
- Glycopeptide profiling
- Native MS characterization
- Middle-down hinge-region analysis
Primary Goal:
- Deliver complete fingerprint-like characterization demonstrating no clinically meaningful differences from the reference biologic
Discover how to study intact proteins in their biologically active states using Native Mass Spectrometry for Biosimilars.
Conclusion: No Single Proteomics Strategy Is Universally Superior for Biosimilars
Framing the discussion as “top-down versus bottom-up” oversimplifies the analytical realities of biosimilar characterization. The more meaningful question is which combination of proteomics approaches can collectively answer the full spectrum of analytical and regulatory questions required for biosimilarity assessment.
Bottom-up proteomics continues to serve as the analytical backbone of biosimilar characterization due to its unmatched sequence coverage, PTM localization capabilities, throughput, and extensive regulatory precedent.
Top-down proteomics contributes critical proteoform-level insight, particularly for intact mass confirmation, charge variant assignment, and holistic structural integrity evaluation.
Middle-down proteomics is rapidly establishing itself as a valuable bridge strategy, especially for hinge-region characterization and large-fragment PTM analysis.
At ResolveMass Laboratories Inc., biosimilar characterization programs are designed using a phase-appropriate, risk-based analytical framework that integrates complementary proteomics approaches to generate the comprehensive molecular evidence expected by regulatory authorities and required for efficient biosimilar advancement.
📩 Ready to discuss your biosimilar characterization strategy?
Contact ResolveMass Laboratories Inc.
Frequently Asked Questions (FAQs)
Top-down proteomics cannot entirely replace bottom-up proteomics in biosimilar analysis because both techniques provide different but equally important analytical information. Top-down methods are highly effective for evaluating intact proteoforms, molecular heterogeneity, and overall structural integrity. However, they do not consistently provide the detailed sequence coverage and precise PTM site localization achievable with bottom-up peptide mapping. Regulatory authorities generally expect a combination of orthogonal analytical techniques, making bottom-up proteomics essential for confirming primary structure and site-specific modifications.
Bottom-up proteomics is generally considered the preferred method for detailed glycosylation analysis because glycopeptide LC-MS/MS enables highly accurate site-specific N-glycan characterization. This approach allows precise identification of glycan structures attached to specific amino acid residues, particularly important sites such as Asn-297 in monoclonal antibodies. In contrast, native top-down or intact glycoprotein mass spectrometry offers a broader molecular overview by providing whole-molecule glycoform distribution patterns. In biosimilar development, both approaches are complementary and together provide a more complete glycosylation profile.
Middle-down proteomics differs from top-down analysis because it uses controlled enzymatic digestion to generate large protein fragments instead of analyzing the complete intact protein. These fragments usually range between 3 and 50 kDa, making them easier to analyze while still preserving substantial structural context. In biosimilar characterization, middle-down workflows are especially valuable for hinge-region analysis, Fc glycosylation studies, and charge variant assessment. This approach offers improved analytical tractability compared to full top-down methods while maintaining more molecular context than conventional bottom-up digestion.
Top-down proteomics requires advanced mass spectrometry platforms capable of handling large molecular masses with high resolving power and sophisticated fragmentation capabilities. Commonly used systems in biosimilar laboratories include the Thermo Scientific Orbitrap Astral, Orbitrap Eclipse with ETD functionality, Q Exactive HF-X, Bruker timsTOF SCP, and FT-ICR instruments. These systems are designed to support intact protein or large-fragment analysis with high mass accuracy. For subunit-level characterization following IdeS digestion, modern high-resolution Orbitrap instruments are often sufficient for reliable analysis.
The FDA’s “fingerprint-like characterization” recommendation emphasizes the importance of generating comprehensive analytical evidence through multiple orthogonal methods. This guidance supports the use of both top-down and bottom-up proteomics rather than relying on a single analytical platform. Bottom-up proteomics provides detailed peptide-level information, including sequence confirmation and PTM localization, while top-down analysis contributes intact mass verification and proteoform characterization. Together with biophysical and functional assays, these complementary approaches help establish the totality of evidence required for biosimilar evaluation.
Different PTMs are better suited to different analytical strategies depending on the level of structural information required. Bottom-up proteomics is highly effective for identifying oxidation and deamidation sites because peptide-level analysis provides precise localization with surrounding sequence information. Glycosylation analysis is also commonly performed using bottom-up glycopeptide workflows for site-specific profiling. In contrast, top-down proteomics is particularly advantageous for detecting modifications such as C-terminal lysine clipping and N-terminal pyroglutamate formation directly on intact proteins or large fragments. Additionally, only top-down analysis preserves information about multiple PTMs occurring simultaneously on the same proteoform.
Bottom-up proteomics serves as the central analytical component of most biosimilar BLA submissions because it supports peptide mapping, glycopeptide analysis, and disulfide bond characterization. Nevertheless, it is rarely considered adequate as a standalone strategy for complete biosimilar characterization. Regulatory agencies generally expect additional orthogonal evidence, including intact mass analysis, charge variant characterization, and higher-order structural confirmation. These complementary datasets often require top-down, native MS, or other intact-level analytical methods. Submissions relying solely on bottom-up data may face requests for further analytical clarification during regulatory review.
Enzymatic digestion can sometimes mask structural differences that exist at the intact protein level because the original molecular context is fragmented into smaller peptides. Certain alterations, including large-scale rearrangements, fusion events, or high-molecular-weight aggregates, may not produce detectable peptide signatures in standard tryptic workflows. In addition, sample preparation artifacts such as missed cleavages, non-specific digestion, or artificial deamidation can introduce analytical variability. For this reason, carefully optimized digestion protocols, rigorous controls, and validated workflows are critical for maintaining data reliability in biosimilar comparability studies.
Reference:
- U.S. Food and Drug Administration. (2015). Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. https://www.fda.gov/media/82647/download
- European Medicines Agency. (2014). Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues. EMA/CHMP/BMWP/42832/2005 Rev1.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (1999). ICH Q6B: Specifications: Test procedures and acceptance criteria for biotechnological/biological products. https://database.ich.org/sites/default/files/Q6B%20Guideline.pdf

