
Introduction
This comprehensive research report examines the advanced scientific, aseptic manufacturing, and regulatory strategies necessary for the successful development and commercialization of generic topical ocular therapies. Collaborating with a specialized provider of CDMO Services for Generic Ophthalmic Drug Products is a key strategy that enables pharmaceutical developers to address the complex formulation challenges, sterile processing requirements, and bioequivalence demonstration pathways associated with these products. Fueled by the patent expiries of blockbuster products containing molecules such as latanoprost, bimatoprost, travoprost, and fluoroquinolones, the global generic ophthalmic sector represents an annual market value exceeding USD 6 billion. Nevertheless, demonstrating bioequivalence remains particularly challenging because drug concentrations at local ocular sites of action—including the cornea, aqueous humor, and uveal tissues—are generally inaccessible through conventional plasma pharmacokinetic (PK) sampling. Therefore, establishing therapeutic equivalence requires an advanced combination of physicochemical characterization, non-invasive in vitro release testing, and state-of-the-art sterile manufacturing validation.
Explore solutions to compress your timelines: Learn how an experienced partner can accelerate generic drug development across the US and Canada to capture market opportunities efficiently.
Share via:
Article Summary:
- Generic ophthalmic drug development is technically complex because products must meet strict requirements for formulation similarity, sterile manufacturing, bioequivalence, and packaging integrity.
- Advanced formulation deformulation and reverse engineering help identify and quantify excipients, polymers, surfactants, preservatives, and other formulation components in the reference listed drug to support Q1/Q2 sameness and product development.
- ANDA approval strategies depend on the dosage form, with ophthalmic solutions, suspensions, emulsions, gels, and ointments requiring different combinations of physicochemical characterization, in vitro testing, IVRT, and, where necessary, in vivo bioequivalence studies.
- Critical quality attributes such as particle size, viscosity, rheology, and dissolution behavior are especially important for ophthalmic suspensions because they can influence drug release, physical stability, ocular retention, and therapeutic performance.
- Sterile manufacturing requires robust aseptic controls, including Blow-Fill-Seal technology, contamination control strategies, environmental monitoring, aseptic process simulations, and operator qualification to minimize contamination risks.
- Container closure integrity and packaging compatibility are essential for maintaining sterility and product quality. Deterministic CCIT methods, extractables and leachables testing, and innovative preservative-free multidose systems help protect ophthalmic products throughout their shelf life.
- A specialized CDMO partner can integrate formulation science, analytical testing, sterile manufacturing support, packaging evaluation, and regulatory strategy to reduce development risks, streamline ANDA preparation, and accelerate the commercialization of generic ophthalmic drug products.

Advanced CDMO Services for Generic Ophthalmic Drug Products and Formulation Deformulation
Establishing qualitative and quantitative formulation identity through specialized CDMO Services for Generic Ophthalmic Drug Products represents a fundamental step in securing clinical study biowaivers and accelerating product development timelines. Formulation deformulation, also referred to as reverse engineering, involves the application of advanced analytical techniques to extract, identify, and quantify all inactive ingredients (excipients) present in a reference listed drug (RLD). This analytical procedure is highly complex because ophthalmic matrices frequently contain trace-level functional polymers, surfactants, and preservatives that must be accurately characterized and mapped to meet regulatory submission requirements.
Optimize your supply chain strategy: Discover the strategic benefits of outsourcing generic drug development in Canada to navigate complex formulations.
To characterize these complex formulations, developers utilize advanced analytical workflows:
Polymer and Viscosity Agent Deformulation: Viscosity-modifying agents, including carbomers, hyaluronates, and cellulosics, are characterized using high-resolution nuclear magnetic resonance (NMR) spectroscopy and Gel Permeation Chromatography (GPC) to determine molecular weight distributions and copolymer ratios.
Surfactant and Micelle Characterization: Trace concentrations of non-ionic surfactants, such as polysorbates and polyoxyl castor oil, are separated and quantified using liquid chromatography-mass spectrometry (LC-MS). This enables developers to reproduce thermodynamic properties and active pharmaceutical ingredient (API) solubilization behavior.
Trace Impurity and Preservative Profiling: Quantitative LC-MS/MS workflows are used to identify and establish the concentration profiles of preservatives, including benzalkonium chloride, while also correlating trace-level degradation pathways to support the evaluation of product shelf life.
This reverse-engineering capability supports a broad range of development objectives. It facilitates generic drug optimization, helps protect intellectual property during disputes, and supports the maintenance of formulation uniformity across commercial manufacturing batches.
Partner with analytical experts: Access advanced testing workflows for analytical development for generic drugs in Canada to de-risk your program early.
Regulatory Framework and ANDA Strategy
The regulatory strategy for obtaining an Abbreviated New Drug Application (ANDA) approval for a generic ophthalmic drug is primarily determined by the physical state of the dosage form and the degree of formulation alignment with the reference listed drug. Under the Federal Food, Drug, and Cosmetic Act (FD&C Act) and 21 CFR Part 314, generic drug products must demonstrate therapeutic equivalence to their RLD. This requires the product to establish both pharmaceutical equivalence—meaning that it contains the same active ingredients, dosage form, strength, and route of administration—and bioequivalence.
| Dosage Form Class | Key Physical State | Inactive Ingredient Criteria (21 CFR 314.94) | Recommended Bioequivalence Pathway |
|---|---|---|---|
| Topical Ophthalmic Solutions | Simple aqueous liquid | Must be qualitatively (Q1) and quantitatively (Q2) identical to the RLD, except for permitted exception excipients. | In vitro biowaiver under 21 CFR 320.22(b), supported by physical-chemical characterization. |
| Topical Ophthalmic Suspensions | Dispersed microparticulate solid in liquid | Q1 and Q2 identity is highly preferred; microstructural (Q3) parameters must also be appropriately matched. | Tiered in vitro studies, including particle size, rheology, and dissolution, or in vivo aqueous humor PK. |
| Topical Ophthalmic Emulsions | Liquid-in-liquid micro-dispersed lipid phase | Strict Q1/Q2 sameness is required, along with extensive matching of globule size distribution. | Multi-pronged in vitro characterization combined with highly sensitive in vitro release testing (IVRT). |
| Topical Ophthalmic Gels & Ointments | Viscoelastic semi-solid matrix | Q1 and Q2 matching is critical and requires replication of complex polymer networks. | In vitro comparative characterization and validated IVRT studies under USP. |
Navigate complex filing requirements seamlessly: Ensure strict compliance with robust regulatory support for generic drugs via a US and Canada CDMO partner.
Evaluating Q1 and Q2 Sameness Under 21 CFR 314.94
Demonstrating qualitative (Q1) and quantitative (Q2) formulation sameness requires evidence that the generic candidate contains the same components at the same concentrations, within the permitted tolerance of ±5%, as the reference listed drug. Under 21 CFR 314.94(a)(9)(iv), the USFDA allows exceptions for certain inactive ingredients, including preservatives, buffers, substances used to adjust tonicity, and thickening agents, provided that the applicant appropriately identifies and characterizes the differences. However, the regulatory agency cautions that any qualitative or quantitative variation in these exception excipients may influence ocular drug absorption or cause corneal irritation. Such differences may consequently necessitate in vivo bioequivalence testing.
Comparative Global Ophthalmic Bioequivalence Guidelines: FDA SGE versus EMA Frameworks
The USFDA and European Medicines Agency (EMA) regulate ophthalmic bioequivalence through distinct and highly structured regulatory pathways. The USFDA implements its requirements through its Product-Specific Guidance (PSG) database, which includes more than 80 ophthalmic reference products and defines clear, tiered approaches for demonstrating bioequivalence. In contrast, the EMA applies its product-specific bioequivalence guideline (PSBGL) framework together with the general “Guideline on Investigation of Bioequivalence” (CPMP/EWP/QWP/1401/98 Rev. 1). Importantly, generic sponsors should recognize that the EMA’s “Guideline on Quality and Equivalence of Locally Applied, Locally Acting Cutaneous Products” (EMA/CHMP/QWP/708282/2018), which became effective in April 2025, applies specifically to skin preparations and does not represent mandatory guidance for eye drops. Nevertheless, its principles for microstructural comparison may be considered by analogy where scientifically appropriate.
Streamline your cross-border submissions: Choose an expert CDMO for generic drug development in Canada to hit your North American milestones.
Advanced Formulation and Manufacturing Science for Ophthalmic Suspensions
The successful manufacturing of ophthalmic suspensions depends on the precise control of critical quality attributes, including particle size distribution and rheology, through advanced milling technologies and polymeric stabilization strategies. Because eye drops are rapidly cleared following administration through tear turnover and blink-induced drainage, the suspension must be designed to provide predictable active ingredient release while minimizing the potential for ocular surface irritation.
Optimizing Critical Quality Attributes via CDMO Services for Generic Ophthalmic Drug Products
The physical stability, ocular retention, and in vivo dissolution kinetics of an ophthalmic suspension are primarily influenced by the drug particle size distribution (PSD) and dispersion viscosity. Working with a specialized provider of CDMO Services for Generic Ophthalmic Drug Products enables these critical attributes to be optimized simultaneously. Following topical instillation, suspended drug particles dissolve in the tear fluid at a rate that is directly related to their total surface area, which is determined by the D50 and SPAN values:
SPAN = (D90 − D10) / D50
To demonstrate bioequivalence, the generic formulation must reproduce the D50 and SPAN profiles of the reference listed drug. These characteristics are evaluated using population bioequivalence (PBE) statistical analysis across multiple commercial-scale batches.
Bring complex pipelines to life: Leverage a specialized CDMO for generic projects in Canada equipped for advanced microstructural characterization.
Advanced Milling, Homogenization, and Polymer Interaction Dynamics
The technical execution of particulate milling requires the selection of a highly reproducible homogenization technology capable of producing a narrow particle size distribution without introducing chemical impurities or causing thermal degradation. FDA research comparing probe sonication, microfluidization, and planetary centrifugal media milling has demonstrated that planetary centrifugal media milling can produce the narrowest and most stable PSD profiles for ophthalmic suspensions.
Generic developers must optimize these milling processes through systematic experimental designs, including central-composite or Box-Behnken methodologies, to evaluate the interactions between critical process parameters and formulation variables:
Milling Kinetic Parameters: Grinding bead size, orbital agitating intensity, and total process run-time must be carefully balanced to regulate the extent of particulate breakdown.
Homogenization Variables: The shear rate and shear time applied during homogenization directly affect the structural assembly and organization of the viscosity-enhancing polymer.
Polymeric and Electrolytic Stabilization: The concentration of viscosity agents, such as Carbomer, must be appropriately coordinated with electrolytes, including sodium chloride. Sodium chloride can exert a significant effect on Carbomer-based formulations because sodium ions shield the carboxylic acid charges along the polymer backbone. This interaction promotes conformational collapse and subsequently reduces viscosity.
Particulate-Polymer Absorption: The physical stability of the suspension depends on molecular-level interactions in which the polymer adsorbs onto the solid API particles. This process creates a protective steric barrier that reduces flocculation and modifies the apparent rheological profile of the suspension.
Scale up with technical confidence: Find end-to-end support with a dedicated generic pharmaceutical CDMO in Canada tailored to complex formulations.
In Vitro Performance Evaluation: IVRT and Advanced Dissolution Testing
In vitro performance testing functions as a highly sensitive and predictive analytical bridge for demonstrating microstructural sameness and determining whether differences in the manufacturing process influence the rate of drug release. For complex generic ophthalmic formulations, including gels, ointments, and suspensions, conventional chemical characterization alone is not sufficient to establish bioequivalence. Therefore, regulatory agencies use in vitro release testing (IVRT) and specialized dissolution profiling to assess product performance under conditions designed to simulate relevant physiological environments.
Access world-class manufacturing expertise: Connect with a top-tier pharmaceutical CDMO in the US and Canada to elevate your product performance evaluations.
Executing USP and Diffusion Cell Methodology
USP General Chapter outlines the accepted technical approaches and apparatus configurations used to determine drug release rates from topical semi-solids and liquid formulations. The most widely accepted system is the Static Vertical Diffusion Cell (VDC), which is commonly known as a Franz cell. The VDC assembly contains a donor chamber loaded with a thick, pseudo-infinite dose of the formulation. This chamber is separated from the receptor chamber by an inert and highly permeable synthetic membrane, such as mixed cellulose esters, polysulfone, or polyethersulfone.
Alternative regulatory-ready apparatus configurations may be selected according to the specific physical characteristics of the dosage form:
USP Apparatus II with Immersion Cell (Model B): This configuration incorporates a 0.53 mL immersion cell positioned inside a flat-bottom vessel. It is particularly appropriate for assessing drug release from more viscous hydrogels and creams.
USP Apparatus IV with Semisolid Adapter: This flow-through cell configuration uses a 0.4 mL semisolid adapter and maintains a continuous flow of receptor solution. This provides high sensitivity for formulations containing low drug doses.
USP Apparatus I with Two-Sided Semisolid Adapter: By placing membranes on both sides of the sample compartment, this configuration substantially increases the available active surface area. As a result, it can improve the discrimination of hydrophobic ophthalmic ointments containing low concentrations of drug.
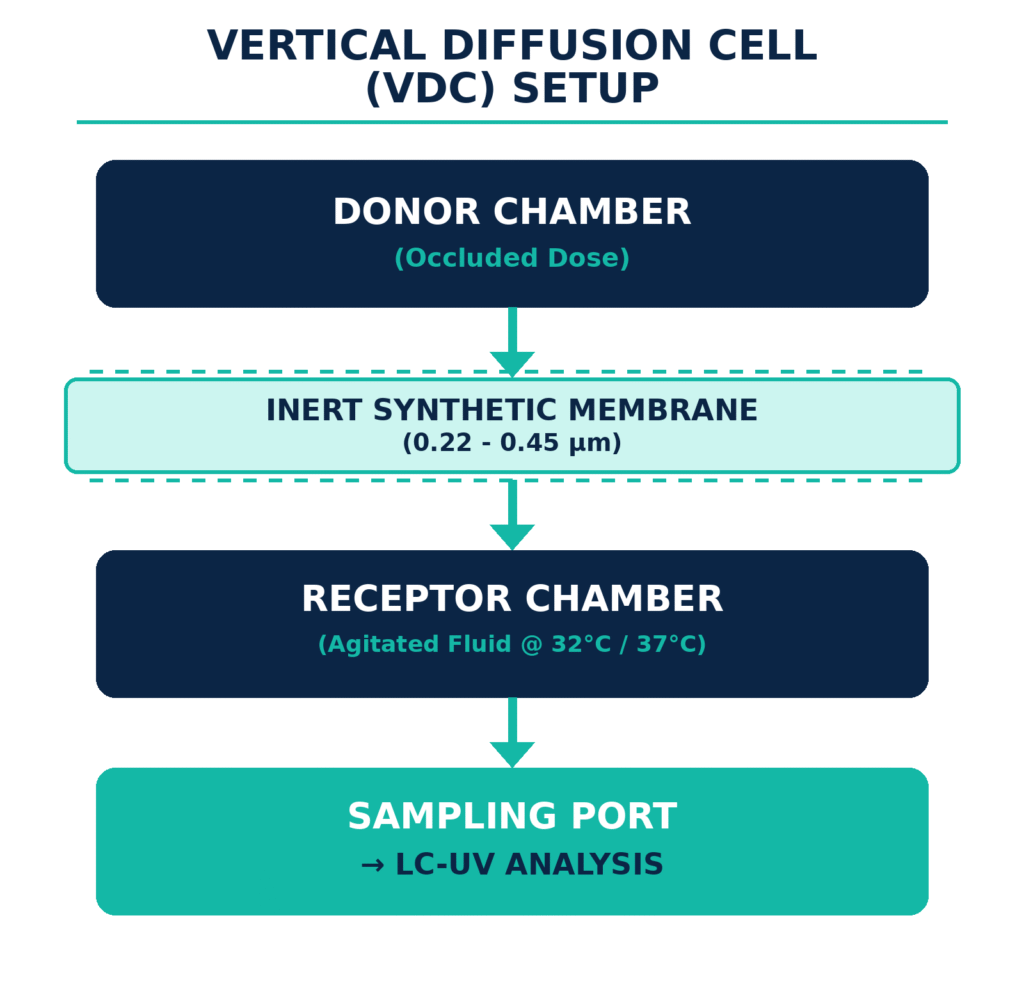
To establish steady-state diffusion kinetics, the drug release profile is typically monitored over a 4- to 6-hour period. During this analysis, the cumulative quantity of drug released (m, expressed in μg/cm²) is plotted against the square root of time (√t). The slope generated from the resulting linear regression represents the in vitro release rate (IVRR).
Discriminatory Dissolution Systems: Simulated Blinking and Fiber Optic UV Testing
Assessing drug release from ophthalmic suspensions requires specialized dissolution approaches capable of capturing rapid dissolution behavior under physiologically relevant stress conditions. Conventional USP dissolution testing generally uses large fluid volumes that do not adequately reproduce the specialized microenvironment of the human eye. To address this limitation, researchers have developed discriminatory dissolution systems based on in-situ fiber optic UV dissolution apparatus.
This advanced testing platform is designed to reproduce important environmental conditions associated with the ocular surface:
Non-Sink Dissolution Dynamics: Testing is performed using micro-volumes of simulated tear fluid (STF, pH 7.4), in which drug concentration can rapidly approach saturation. This reflects the restricted fluid volume available within the tear film.
High-Shear Blinking Replicas: The system introduces rapid mechanical agitation to reproduce the intense shear forces that suspended drug particles experience during human blinking.
Real-Time Data Acquisition: In-situ fiber optic probes continuously measure API concentrations, with data acquisition possible as early as 40 seconds after study initiation. This produces a highly sensitive dissolution profile capable of distinguishing relatively small differences in particle size and polymer structure.
Sterile Processing, Aseptic Fill-Finish, and Contamination Control
The manufacture of sterile ophthalmic preparations requires rigorous environmental controls and highly automated processing systems to minimize the risk of microbial and particulate contamination. Because topically administered eye drops come into direct contact with compromised or sensitive ocular tissues, even low levels of contamination can result in serious clinical consequences, including keratitis and vision loss. For this reason, global regulatory authorities have strengthened manufacturing expectations and placed sterile processing under heightened regulatory scrutiny.
Blow-Fill-Seal vs. Traditional Aseptic Lines under EU GMP Annex 1
The 2022 revision of EU GMP Annex 1 formally recognizes Blow-Fill-Seal (BFS) technology as an advanced aseptic processing method that can substantially reduce contamination risks by minimizing human intervention during critical filling operations. Conventional filling lines require pre-formed glass or plastic vials to undergo sequential washing, sterilization, depyrogenation, transport, filling, and stoppering operations. Each individual transition creates a potential environmental interface through which airborne particulates or human operators may introduce microbial contamination.
By comparison, BFS systems combine container extrusion, filling, and sealing within one automated manufacturing sequence:
Extrusion Phase: Medical-grade thermoplastic polymer resin, such as LDPE, is extruded at temperatures ranging from 170°C to 220°C. The high processing temperature provides sterilization of the resin during its residence time within the extruder.
Molding Phase: The molten polymer tube, known as the parison, is enclosed within a sterile mold, and its lower section is pinched closed to create the body of the container.
Blowing & Filling Phase: The newly formed container is positioned beneath an ISO 5 (Class A) filling zone. Sterile blowing and filling nozzles descend into position, inflate the container using sterile air, and immediately dispense the sterile liquid formulation.
Hermetic Sealing Phase: The sealing mold closes to create the top cap and hermetically seals the container within seconds, thereby avoiding prolonged exposure to the surrounding environment.
| Process Variable | Blow-Fill-Seal (BFS) Technology | Traditional Aseptic Glass/Vial Filling |
|---|---|---|
| Sterility Assurance Level (SAL) | Provides inherently higher sterility assurance through the integrated manufacturing process. | Highly dependent on cleanroom conditions, process controls, and operator technique. |
| Environmental Exposure | Operates as an enclosed system in which the container does not exist as an open vessel during critical processing. | The pre-formed container remains open during transport and filling operations. |
| Human Intervention | Human intervention is eliminated from the critical filling zone. | Human involvement is required for equipment setup, stoppering, and capping adjustments. |
| Supply Chain Complexity | Containers are produced directly from raw polymer granulates during the manufacturing process. | Requires the management of separate vials, rubber stoppers, and aluminum caps. |
| Particulate Levels | Intrinsic and extrinsic particulate counts can be up to 10-fold lower. | Greater potential for glass particle generation and elastomeric shedding. |
Designing and Validating a Sterile Contamination Control Strategy
To achieve audit readiness under Annex 1, sterile manufacturers must establish a site-wide Contamination Control Strategy (CCS) that functions as a dynamic quality document. Instead of managing contamination controls as isolated operational procedures, the CCS applies Quality Risk Management (QRM) principles to connect cleanroom design, HVAC performance, airlock pressure differentials, and raw material controls within a unified compliance framework.
Validation of the sterile manufacturing process requires a comprehensive, multi-dimensional approach:
Aseptic Process Simulations (APS): Commonly known as media fills, these simulations must incorporate worst-case production conditions, including maximum line speeds, multiple shift changeovers, and frequent operator interventions. The objective is to demonstrate the overall process capability and robustness of the aseptic filling line under challenging operating conditions.
Environmental Monitoring (EM): Continuous monitoring of viable and non-viable particulates must be performed in Grade A and B zones during active production. Validated air samplers, contact plates, and settle plates are used to detect contamination events and identify emerging contamination trends.
Personnel Qualification: Operators must receive rigorous and periodic training in aseptic techniques and gowning qualification. This process includes microbiological monitoring of sterile garments to verify that personnel can correctly don protective clothing without introducing external micro-organisms into the controlled manufacturing environment.
Primary Packaging Integrity and Material Science
The container closure system (CCS) for an ophthalmic formulation must be designed to maintain an effective microbial barrier, prevent physical product leakage, and protect the active formulation from environmental degradation. Ophthalmic packaging configurations include single-dose BFS ampoules, conventional multi-dose glass or plastic vials, and sophisticated mechanical multidose preservative-free squeeze bottles. Validation of these different packaging systems requires the integration of deterministic leak testing with comprehensive chemical compatibility assessments.
Deterministic Container Closure Integrity Testing (CCIT) and USP
USP General Chapter establishes the scientific and regulatory framework for assessing container closure integrity (CCI) across sterile pharmaceutical packaging systems. The standard specifically favors deterministic physical leak testing methods because they generate objective, quantitative, and highly reproducible results based on measurable physical properties. These methods are preferred over visual and highly subjective probabilistic approaches, such as liquid dye ingress or microbial challenge testing.
The deterministic methodologies commonly used for ophthalmic container evaluation include:
Vacuum Decay Testing (ASTM F2338): The sealed container is positioned inside a closely fitting evacuation chamber. After the chamber is evacuated, the vacuum source is isolated. A rise in dead-space pressure, indicating vacuum decay, above a predefined limit confirms the presence of a physical defect or crack. This non-destructive method is particularly suitable for conventional plastic and glass vials, BFS strips, and multi-dose bottles.
Helium Mass Spectrometry (Tracer Gas, Vacuum Mode): Helium is introduced into the headspace of the container, after which the package is placed inside an evacuated test chamber. A mass spectrometer measures the rate at which helium escapes from the container. This method is regarded as one of the most sensitive technologies for leak detection and can identify sub-micron defects smaller than 0.01 μm while supporting the establishment of the package’s Maximum Allowable Leakage Limit (MALL).
High-Voltage Leak Detection (HVLD): A high-voltage, high-frequency, low-amperage electrical current is applied across the surface of a liquid-filled container. A leak path containing conductive liquid formulation produces an increase in electrical conductivity compared with an intact container. This non-destructive technique is particularly suitable for liquid-filled parenterals and polymer-based containers.
Laser-Based Headspace Analysis: This frequency-modulated near-infrared spectroscopy technique directs a laser diode through the gas headspace of a sealed, transparent container. It detects changes in headspace oxygen, water vapor, or total absolute pressure, making the technique highly effective for confirming vacuum maintenance and evaluating oxygen-sensitive formulations.
Establishing the MALL is a critical component of a validated CCIT program. It represents the highest leakage rate that can be tolerated for a specific product-package combination without compromising sterility or physicochemical quality throughout the product shelf life. For rigid containers in which product sterility and headspace conditions must be maintained, USP cites a MALL of less than 6 × 10⁻⁶ mbar·L/s as the benchmark standard.
Extractables and Leachables Programs: USP <661.1>, USP <661.2>, and USP <1663> / <1664>
Because topical ophthalmic formulations are commonly packaged in squeezable polymer containers, including low-density polyethylene, they may be particularly susceptible to chemical migration from packaging constituents. Under USP and related requirements, CDMOs must establish an extractables and leachables (E&L) program to identify and control foreign chemical impurities that may migrate from primary, secondary, or tertiary packaging materials.
The structural and chemical evaluation of polymeric packaging components follows a strict, multi-tiered testing strategy:
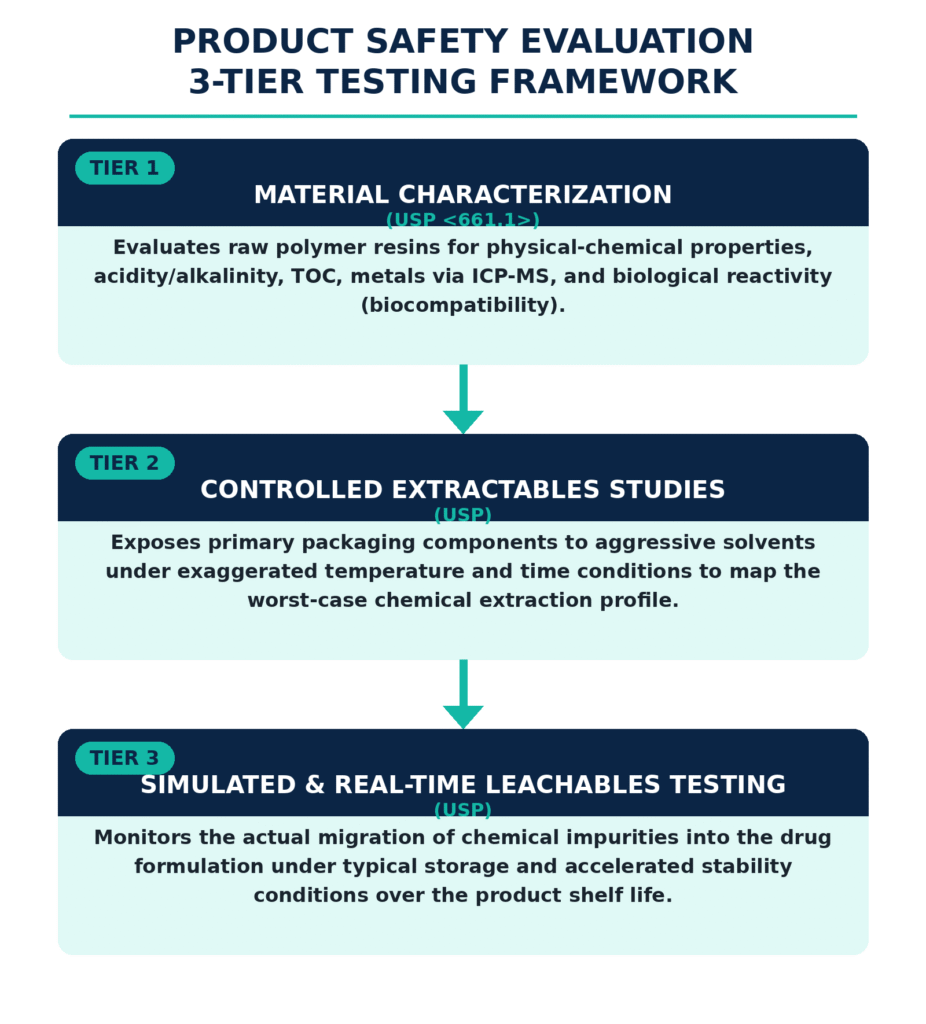
To assess the toxicological risk associated with migrating compounds, developers use the Safety Concern Threshold (SCT). For parenteral and ophthalmic dosage forms, the SCT represents a daily exposure level of 1.5 μg/day below which a leachable is generally considered unlikely to present a significant toxicological or mutagenic concern. The Analytical Evaluation Threshold (AET) is then mathematically derived from the SCT to establish the minimum analytical reporting limit for chromatographic screening:
AET = (SCT / Maximum Daily Dose) × (1 / Analytical Uncertainty Factor)
Common leachables identified in semi-permeable LDPE systems include polymer degradation products, plasticizers, slip agents, antioxidants, and volatile monomers. In addition, volatile substances originating from external labels, printing inks, and adhesive resins may migrate through the semi-permeable plastic container wall. Consequently, a comprehensive assessment of the complete packaging system is required.
De-risk your primary packaging matrices: Deploy validation setups utilizing peptide analytical characterization services to screen structural interactions and material trace leaching profiles.
Packaging Innovations: Multidose Preservative-Free (MDPF) Systems
Multidose preservative-free (MDPF) delivery systems represent an important innovation in ophthalmic packaging. These systems use mechanical barrier designs to prevent microbial contamination during repeated use while eliminating the need for potentially cytotoxic preservatives such as benzalkonium chloride (BAK). Preservatives used in chronic ophthalmic therapies, including glaucoma and dry eye treatments, have been associated with corneal surface toxicity, cellular apoptosis, and patient discomfort.
To preserve formulation sterility throughout multi-week patient-use cycles without relying on chemical preservatives, MDPF systems primarily use two mechanical design approaches:
Tip-Seal Technology (Aptar OSD): The Aptar OSD platform incorporates a mechanical, spring-loaded tip-seal valve together with a metal-free fluid path. When the patient squeezes the bottle, internal pressure causes the valve to open and dispense a calibrated drop. Once the pressure is released, the spring-loaded mechanism immediately reseals the orifice, preventing the backflow of contaminated liquid or lachrymal fluid into the container.
Non-Return Valves with Venting Membranes (Nemera Novelia): The Nemera Novelia platform combines a non-return valve with a solid, non-porous silicone air-compensation membrane based on PureFlow technology. Because the solid silicone membrane contains no physical pores, it uses carefully controlled polymer permeability characteristics to allow sterile air to enter the bottle while maintaining a continuous physical barrier against bacterial and fungal contamination.
Furthermore, the Novelia design is engineered to maintain a high degree of mechanical consistency, with only a 6% increase in the required actuation force between the first and last dose. This compares with an increase of approximately 35% observed in alternative MDPF systems. Such mechanical consistency supports dosing accuracy and patient compliance, particularly among elderly patients and individuals with reduced hand dexterity.
De-risking Development with a Specialized Canadian Partner
ResolveMass Laboratories Inc. is an industry-leading, Health Canada licensed, and FDA-registered Contract Research Organization (CRO) and Contract Development and Manufacturing Organization (CDMO) that provides specialized R&D, advanced material characterization, and GMP testing services. Holding a Drug Establishment Licence (GMP Compliant, Licence 3-002945-A) and maintaining USFDA registration (Establishment Identifier 3042696771), ResolveMass operates under a fully implemented, ISO 9001:2015 certified Quality Management System designed to support data integrity, regulatory compliance, and scientific rigor.
The laboratory provides specialized scientific support designed to reduce risk throughout generic ophthalmic development programs:
Polymer Informatics and Controlled Release Experts: ResolveMass specializes in the custom synthesis and comprehensive analytical characterization of biocompatible polymers, including PLGA, PLA, and PLCL. These materials are important for the development of long-acting ophthalmic injectables and implants. By integrating advanced machine learning algorithms into polymer chemistry, ResolveMass accelerates polymer design and reduces the risks associated with replicating complex RLD controlled-release matrices.
State-of-the-Art Mass Spectrometry and NMR: ResolveMass’s PhD-level scientific team addresses complex analytical challenges, including high-resolution LC-MS/MS impurity profiling, unknown compound structure elucidation, and trace-level nitrosamine and PFAS testing in complex drug matrices.
Comprehensive E&L Program Execution: The laboratory designs and conducts compliant extractables and leachables studies to identify trace contaminants in polymer-based primary packaging and semi-permeable squeeze bottles, supporting compliance with USP and relevant regulatory submission requirements.
Conducting complex analytical characterization and formulation workflows within a single integrated quality system eliminates the inefficiencies associated with multiple vendor handoffs. This streamlined approach can shorten development timelines and accelerate speed-to-market for generic drug developers.
Choose the right operational model: Understand vendor capabilities by reviewing the trade-offs of a CDMO vs CRO for generic drug development to optimize your program’s structure.
Conclusion
In summary, the successful development and commercialization of generic ophthalmic formulations require specialized CDMO Services for Generic Ophthalmic Drug Products capable of addressing complex formulation deformulation, supporting regulatory biowaiver strategies, and implementing compliant aseptic processing systems. By systematically matching qualitative, quantitative, and microstructural (Q1/Q2/Q3) parameters, generic sponsors can address the stringent expectations outlined in FDA Product-Specific Guidances and EMA frameworks without automatically relying on costly clinical trials. The implementation of deterministic container closure integrity testing, together with the verification of sterile processing through a comprehensive, site-wide Contamination Control Strategy, supports long-term product quality and patient safety. Partnering with a specialized, GMP-compliant laboratory such as ResolveMass Laboratories Inc. provides robust data integrity and the advanced analytical infrastructure required to support successful ANDA approvals. For specialized technical support or to initiate a new generic development project, pharmaceutical developers can consult a pharmaceutical scientist directly through the ResolveMass Contact Us page.
Frequently Asked Questions (FAQs)
Yes, 21 CFR 314.94(a)(9)(iv) permits certain differences involving designated “exception excipients,” including preservatives, buffers, substances used to adjust tonicity, and thickening agents. However, the applicant must adequately characterize these differences and demonstrate that they do not negatively influence product safety, drug performance, or therapeutic effectiveness. Changes outside these permitted categories, or differences that alter drug solubility or corneal permeability, may prevent eligibility for a biowaiver and could require in vivo bioequivalence studies.
Q1, Q2, and Q3 sameness describe progressively more detailed levels of equivalence between a generic product and its RLD. Q1 sameness confirms that the same active and inactive ingredients are present, whereas Q2 sameness confirms that these components are present at the same concentrations. Q3 sameness goes further by examining the physical, microstructural, and thermodynamic characteristics of the dosage form, including particle size distribution, shear-dependent viscosity, polymorphic form, and drug distribution within dispersed phases, all of which can influence drug release and in vivo performance.
Planetary centrifugal media milling is often preferred because it can generate a narrow and reproducible particle size distribution (PSD) while maintaining consistent D50 values and limiting thermal stress. The process uses combined rotational and orbital motion to produce intense milling forces through micro-sized grinding media. Compared with techniques such as probe sonication or microfluidization, this approach can reduce broad particle size variability and localized heat generation, supporting improved physical stability and more consistent dosing performance.
Sodium chloride can substantially alter the rheological behavior of carbomer-based ophthalmic suspensions by introducing sodium and chloride ions that screen the electrostatic charges along the acrylic acid polymer backbone. This reduces electrostatic repulsion and compresses the polymer’s electrical double layer, causing the extended polymer network to contract into a more coiled and lower-viscosity configuration. Therefore, electrolyte concentration must be carefully controlled during formulation and processing to prevent unexpected changes in viscosity and sedimentation behavior.
A validated USP Vertical Diffusion Cell (VDC) study requires careful control of several critical parameters, including the selection of an inert synthetic membrane, such as 0.45 μm polyethersulfone, and maintenance of appropriate sink conditions in the receptor medium. The membrane surface temperature should generally be controlled at 32 ± 1°C, or 37 ± 1°C for relevant internal products. Additional requirements include degassing the receptor medium to minimize air bubbles and applying a sufficiently thick, occluded dose to support steady-state release kinetics.
Blow-Fill-Seal (BFS) technology enhances sterility assurance by combining container formation, sterile liquid filling, and hermetic sealing within a single automated and continuous closed-system process. In contrast, conventional aseptic filling exposes pre-formed containers to multiple handling and transfer steps during washing, depyrogenation, transport, filling, and capping. In BFS manufacturing, the polymer container is formed at elevated temperatures of approximately 170–220°C and filled immediately under an ISO 5 (Class A) environment, substantially reducing human intervention in the critical filling zone.
The Maximum Allowable Leakage Limit (MALL) represents the highest leakage rate that a particular sterile product and container closure system can tolerate without compromising sterility, product safety, or physicochemical quality throughout the shelf life. For rigid sterile pharmaceutical containers in which headspace gases or sterility must be maintained, USP cites a benchmark MALL of less than 6 × 10⁻⁶ mbar·L/s. This limit is commonly evaluated using highly sensitive deterministic methods, including vacuum-mode helium mass spectrometry.
Liquid dye ingress is classified as a probabilistic leak detection method because successful detection depends on several variable events occurring together, including liquid movement through a potential capillary pathway, dye penetration under a pressure or vacuum differential, and visual identification by an operator. The method is destructive and may be influenced by subjective interpretation. It also has limited reliability for consistently identifying extremely small cracks or defects compared with quantitative, non-destructive deterministic techniques such as vacuum decay and high-voltage leak detection.
The Analytical Evaluation Threshold (AET) supports risk-based E&L testing by defining a scientifically and mathematically justified concentration above which migrating chemical substances must be identified and evaluated toxicologically. The value is derived using the Safety Concern Threshold (SCT), the product’s maximum daily dose, and an analytical uncertainty factor. For high-risk ophthalmic and parenteral products, an SCT of 1.5 μg/day is commonly applied, helping ensure that trace polymer degradants, plasticizers, and contaminants from packaging materials are not overlooked.
Reference:
- Choi, S. H., & Lionberger, R. A. (2016). Clinical, pharmacokinetic, and in vitro studies to support bioequivalence of ophthalmic drug products. The AAPS Journal, 18(4), 1032–1038. https://doi.org/10.1208/s12248-016-9932-z
- U.S. Food and Drug Administration. (2025, March 21). Helpful webinars and other resources for generic drug manufacturers. FDA resource
- Myung, J. H. (2022, August 18). Introduction of bioequivalence for generic drug products [PowerPoint slides]. U.S. Food and Drug Administration. FDA presentation
- Beringhs, A. O., Naageshwaran, V., Gum, G., Seremak, D., Malla, S., Vo, A., Tan, M.-L., Babiskin, A., Wang, Y., Xu, X., & Kozak, D. (2023). Impact of variations in critical quality attributes of brinzolamide ophthalmic suspensions on preclinical pharmacokinetics and pharmacodynamics following once-daily topical instillations [Conference poster]. U.S. Food and Drug Administration. https://www.fda.gov/media/168610/download
- U.S. Food and Drug Administration. (2019). FY2018 GDUFA science and research report: Ophthalmic products. https://www.fda.gov/media/130617/download

